Recognize the Early Signs

Recognize the Early Signs
Expedite early diagnosis by recognizing the early hallmark symptoms of CLN2 disease
New-onset, unprovoked seizures
- Unprovoked seizures occur between the ages of 2 and 4 and are the symptom that most often leads parents/caregivers to seek medical attention1,2
- Initial new-onset, unprovoked seizures are commonly generalized tonic-clonic, myoclonic, or atonic2-4
- For some children, the first seizure can be febrile in nature
Early language delay
- Language delay is one of the earliest symptoms to appear according to recent scientific findings5
Percentage of “Late Talkers” in children with CLN2 disease vs age-matched controls (n=36)5
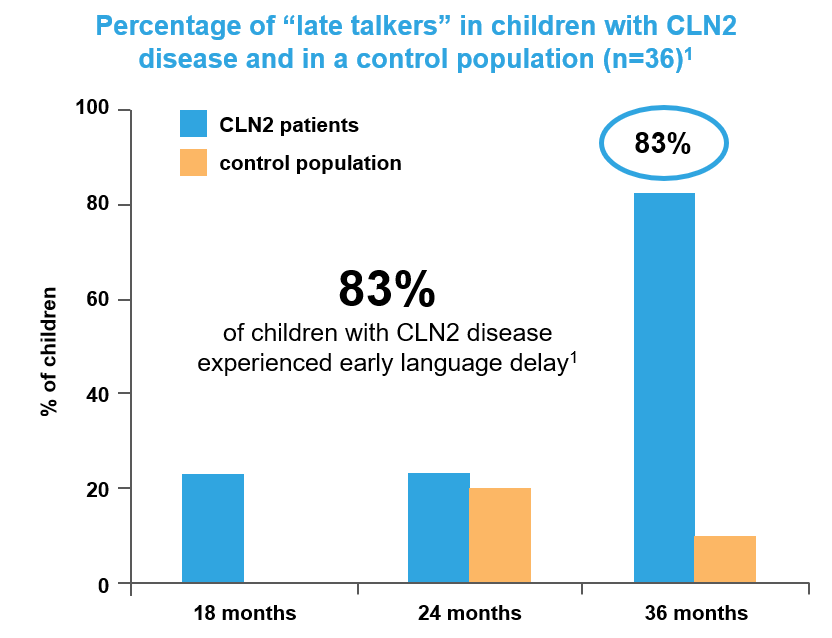
- Delay in early language development was defined by the following criteria5:
- First single words acquired at 18 months (or later/never)
- First two-word sentences at 24 months (or later/never)
- First full sentences at 36 months (or later/never)
- In contrast, a separate study reported that only 27% of children (aged 3 to 5) who presented with unprovoked seizures had a history of language difficulties6
Motor disturbances
- 40% report loss of motor abilities (eg, ataxic gait, clumsiness/tripping, or frequent falls) as the first symptom5
Note: Unlike other NCLs, visual impairment isn't typically present until later stages of the disease, well into the neurodegenerative decline of CLN2 disease.3
Electrophysiological and radiological findings may provide early signals of CLN2 disease

Electroencephalogram (EEG) with low-frequency (1-2 Hz) intermittent photic stimulation (IPS) elicits a distinguishing photoparoxysmal response (PPR) in the majority of children with CLN2 disease7
According to a recent study of children with CLN2 disease, PPR was7:
- Seen in 60% of children
- Identified on the initial EEG in 78% of cases
- Detected in time-locked fashion in 63% of cases

Early in the disease, a brain MRI may appear normal or show subtle abnormalities3,8
Abnormalities include:
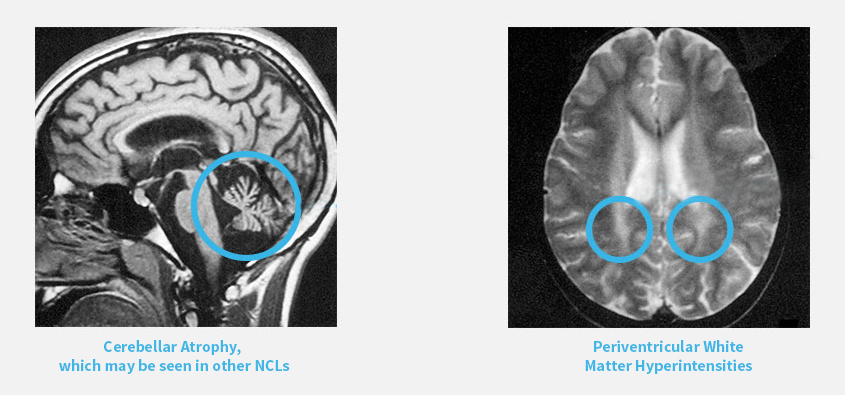
Photos courtesy of Ludovico D’Incerti, MD.
Later in the disease, cerebral atrophy becomes evident.
Not all children with CLN2 disease will exhibit these findings. A definitive diagnosis should be confirmed through laboratory testing.
BioMarin has partnered with Invitae to bring you the Behind the Seizure® program—a no-cost epilepsy gene panel testing program to help you diagnose CLN2 disease early.

In as little as 2 weeks, an epilepsy gene panel test can bring you and your eligible patients closer to identifying if there is a genetic cause behind the seizure.
Visit www.behindtheseizure.com to learn more and to order a test.
The importance of seeing the early signs of CLN2 disease
Dr Angela Schulz, MD, PhD
Children’s Hospital
NCL Specialty Clinic
International Center of Lysosomal Storage Disorders (ICLD)
University Medical center Hamburg-Eppendorf
Hamburg, Germany
Children’s Hospital
NCL Specialty Clinic
International Center of Lysosomal Storage Disorders (ICLD)
University Medical center Hamburg-Eppendorf
Hamburg, Germany
A discussion about the early signs of CLN2 disease (new-onset, unprovoked seizures and language delay) and how earlier diagnosis may benefit the child and their family.
References: 1. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases – clinical perspectives. Biochimica et Biophysica Acta. 2013;1832:1801–1806.
2. Schulz A, Cohen-Pfeffer JL, Crystal R, et al. Neuronal ceroid lipofuscinosis-2 (CLN2) disorder, a type of Batten disease caused by TPP1 enzyme deficiency: current knowledge of the natural history from international experts. Poster session presented at: The Society for the Study of Inborn Errors of Metabolism (SSIEM) Annual Symposium; September 2015; Lyon, France. 3. Chang M, Cooper JD, Davidson BL, et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 4. Pérez-Poyato MS, Marfa MP, Abizanda IF, et al. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. J Child Neurol. 2013;28:470-478. 5.Nickel M, Simonati A, Jacoby D, et al. Disease characteristics and progression in patients with late-infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease: an observational cohort study. Lancet Child Adolesc Health. 2018 Aug;2(8):582-590. doi: 10.1016/S2352-4642(18)30179-2. 6. Åndell E, Tomson T, Carlsson S, et al. The incidence of unprovoked seizures and occurrence of neurodevelopmental comordities in children at the time of their first epileptic seizure and during the subsequent six months. Epilepsy Res. 2015;113:140-150. 7. Albert DV, Yin H, De Los Reyes EC, Vidaurre J. Unique Characteristics of the photoparoxysmal response in patients with neuronal ceroid lipofuscinosis type 2: can EEG be a biomarker? J Child Neurol. 2016;31:1475-1482. 8. Mole SE, Williams RE, and Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126.
