Pathophysiology

Pathophysiology
TPP1 enzyme deficiency results in neurodegeneration in children with CLN2 disease1
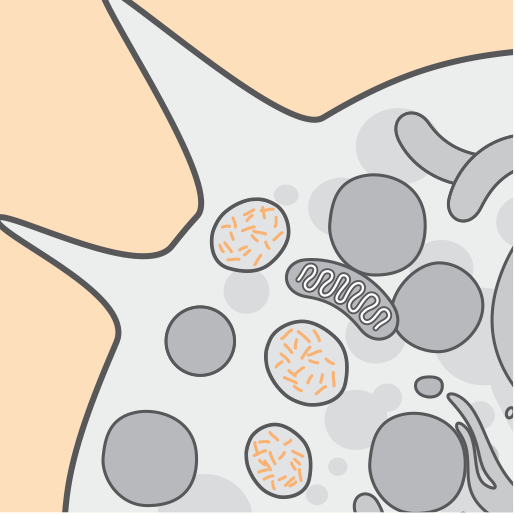
Mutations in the CLN2/TPP1 gene result in deficient activity of the tripeptidyl‐peptidase 1 (TPP1) enzyme. TPP1 is a protease that cleaves N-terminal tripeptides from substrates in lysosomes.1,2
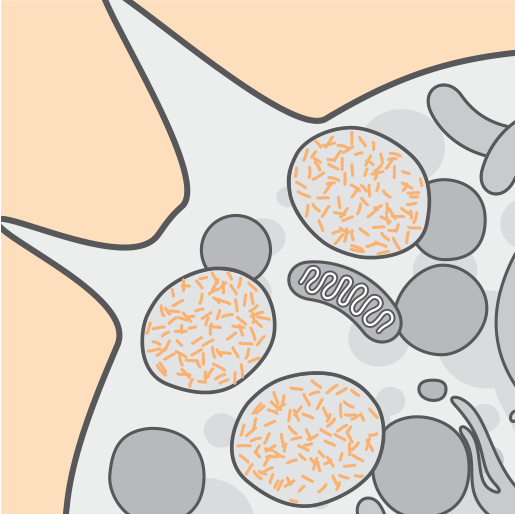
The absence or reduced activity of the TPP1 enzyme is associated with an accumulation of lysosomal autofluroescent lipopigment storage material.2,3
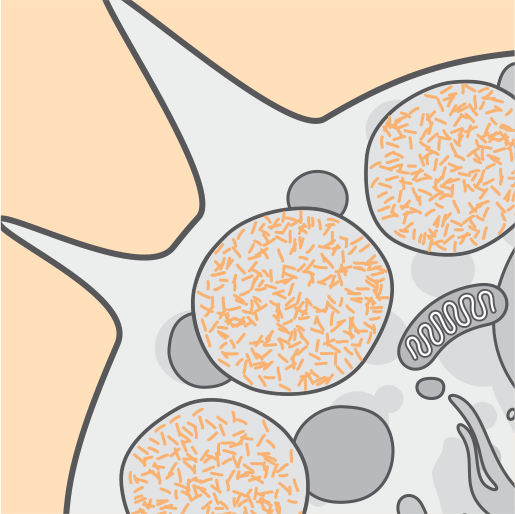
Over time, cell dysfunction, cell death, and atrophy occur.3,4
Continuous accumulation in lysosomes damages cells and the devastating symptoms of CLN2 disease become noticeable.
References: 1. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases – clinical perspectives. Biochimica et Biophysica Acta. 2013;1832:1801-1806. 2. Mole SE, Williams RE, and Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126. 3. Chang M, Cooper JD, Davidson BL, et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 4. Haltia M. The neuronal ceroid-lipofuscinoses: from past to present. Biochimica et Biophysica Acta. 2006;1762:850-856.
