 Español
Español UK (English)
UK (English) Deutsch
Deutsch Italiano
Italiano Русский
Русский Français
Français
Forget your password?
Necesidad de un diagnóstico precoz

Necesidad de un diagnóstico precoz
Las señales y los síntomas de la enfermedad CLN2 van más allá de la epilepsia
A pesar de esto, los niños a menudo son diagnosticados con epilepsia sin un estudio diagnóstico adicional para la enfermedad CLN2
- La edad promedio de diagnóstico es de 5 años – cuando el declive neurodegenerativo de la enfermedad CLN2 ya está bien adelantado.
- Las razones de atraso en el diagnóstico de la enfermedad CLN2 incluyen1,2:
- Los síntomas iniciales son inespecíficos.
- La progresión de la enfermedad CLN2 se puede confundir con los efectos secundarios de los fármacos antiepilépticos.
- El diagnóstico precoz permite el acceso al cuidado clínico y apoyo social específicos para la enfermedad CLN2, lo que puede repercutir positivamente en los resultados y la calidad de vida del niño y de la familia.
- El diagnóstico precoz de la enfermedad CLN2 es crucial para que los padres sepan que son portadores genéticos y asi reforzar la importancia de la planificación familiar.3
- Los retrasos en el diagnóstico pueden ir acompañados de evaluaciones exhaustivas y diagnósticos erróneos antes de confirmar un diagnóstico definitivo.
El diagnóstico de la enfermedad CLN2 se retrasa un promedio de 2 años desde la primera convulsión observada a los 3 años.
La enfermedad CLN2 a menudo no se diagnostica correctamente desde el inicio, lo que retrasa el diagnóstico preciso
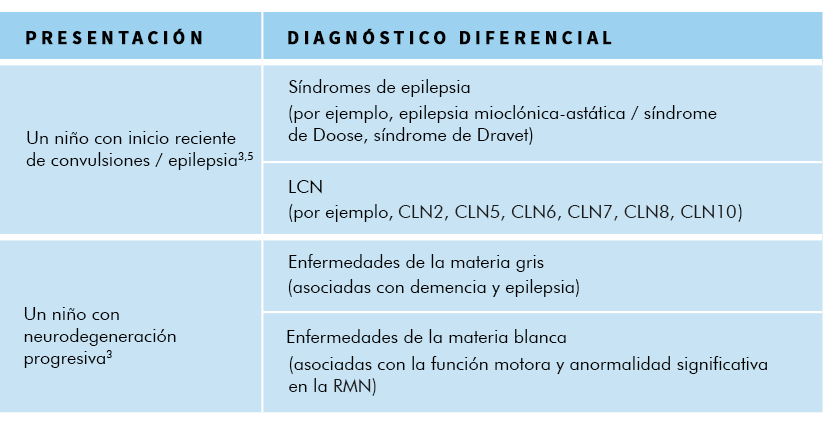
En los inicios de la progresión de la enfermedad CLN23,4:
- Dado que las convulsiones y las mioclonías son el síntoma más prominente, se suelen sospechar síndromes en las que la mioclonía es común (por ejemplo, epilepsia focal, síndrome de epilepsia mioclónica/epilepsias mioclónicas astáticas, como el síndrome de Doose, síndrome de Dravet, síndrome de Lennox-Gastaut, u otros síndromes de epilepsia).
Más tarde en la progresión de la enfermedad CLN23:
- A medida que la enfermedad progresa y la regresión psicomotora y pérdida de la función se hacen más prominentes, se pueden sospechar otros trastornos pediátricos cerebrales progresivos (por ejemplo, inflamación/infecciones, tumores, trastornos mitocondriales y otras enfermedades de depósito lisosomal, incluidos otros tipos de LCN).
Referencias: 1. Fietz M, Giugliani R, AlSayed M, et al. Expert recommendations for the laboratory diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): diagnostic algorithm and best practice guidelines for a timely diagnosis. Poster session presented at: The 12th Annual WORLD Symposium; February – March 2016; San Diego, CA. 2. Williams RE, Adams HR, Blohm M, et al. Expert opinion on the management of CLN2 disease. Poster session presented at: The 12th Annual WORLD Symposium; February – March 2016; San Diego, CA. 3. Chang M, Cooper JD, Davidson BL, et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 4. Pérez-Poyato MS, Marfa MP, Abizanda IF, et al. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. J Child Neurol. 2013;28:470-478. 5. Mole SE, Williams RE, and Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126.
