This site is for healthcare professionals only. Please click here if you are not a healthcare professional.
 UK (English)
UK (English)
 Español
Español Türkçe
Türkçe Deutsch
Deutsch Italiano
Italiano Русский
Русский Français
Français
Forget your password?
Natural history

Natural history
CLN2 disease follows a devastatingly rapid course – symptoms and functional loss progress with age
Timeline of classic CLN2 disease: symptom onset and loss of function1–7

Age ranges depicted are averages for the classic late-infantile phenotype. Atypical phenotypes of CLN2 disease can vary in age of onset, rate of progression and disease manifestation.
Ages 0 to 2
- Lysosomal accumulation of lipopigments is thought to begin in the prenatal period, yet children appear to develop normally until the onset of presenting symptoms4
- The first symptom in the majority of cases is early language delay, as shown in recent clinical findings8
- In a minority of cases, other developmental delays, motor difficulties, or ataxia may be the first sign5,6,9
- Often these can occur together with language delay
Ages 2 to 4
- Even though language delay may appear earlier, seizures (which occur around age 3) are the symptom that most often leads parents to seek medical attention for CLN2 disease4,8
- Multiple types of seizures can be observed, including generalised tonic-clonic, absence, myoclonic, atonic, clonic and tonic4,5
- Although seizures are most often unprovoked, febrile seizures may occur5
- Onset of ataxia often occurs during this stage of the disease6
Ages 4 to 7
- Symptoms compound with age, and there is a dramatic loss of previously attained skills. During this age range, children with CLN2 disease typically experience:2–4,10
- Language regression
- Myoclonic seizures, both epileptic and non-epileptic
- Severe motor function impairment with eventual loss of voluntary movements
- Movement disorders such as myoclonus, spasticity, dystonia and chorea
- Visual deterioration which leads to blindness by age 6 or 7
- Eventually, there is a complete loss of language and ability to walk and children become wheelchair-dependent, bedridden, and completely reliant on carers4
Ages 8 to 12
- CLN2 disease is characterised by premature death and the majority of children rarely survive past early adolescence3
Blindness is an early symptom in other NCLs, but occurs in the later stage of CLN2 disease.2,7

Dr Paul Gissen, MBChB, PhD, FRCPCH
University College London
Great Ormond Street Hospital for Children
NHS Foundation Trust
University College London
Great Ormond Street Hospital for Children
NHS Foundation Trust
An overview of the predictable nature of CLN2 disease and when symptoms develop throughout the lives of affected children.
The CLN2 disease clinical rating scale is an efficient way to assess disease progression in 2 major functional areas: motor and language ability
The standardised scoring system can be used to quantitatively assess disease progression at diagnosis and track loss of function over time7- Each functional area is scored on a scale of 3 (grossly normal) to 0 (profoundly impaired)7
- The highest possible score when assessing motor and language function is 68
Functional performance ability is rated as follows:7,8,10
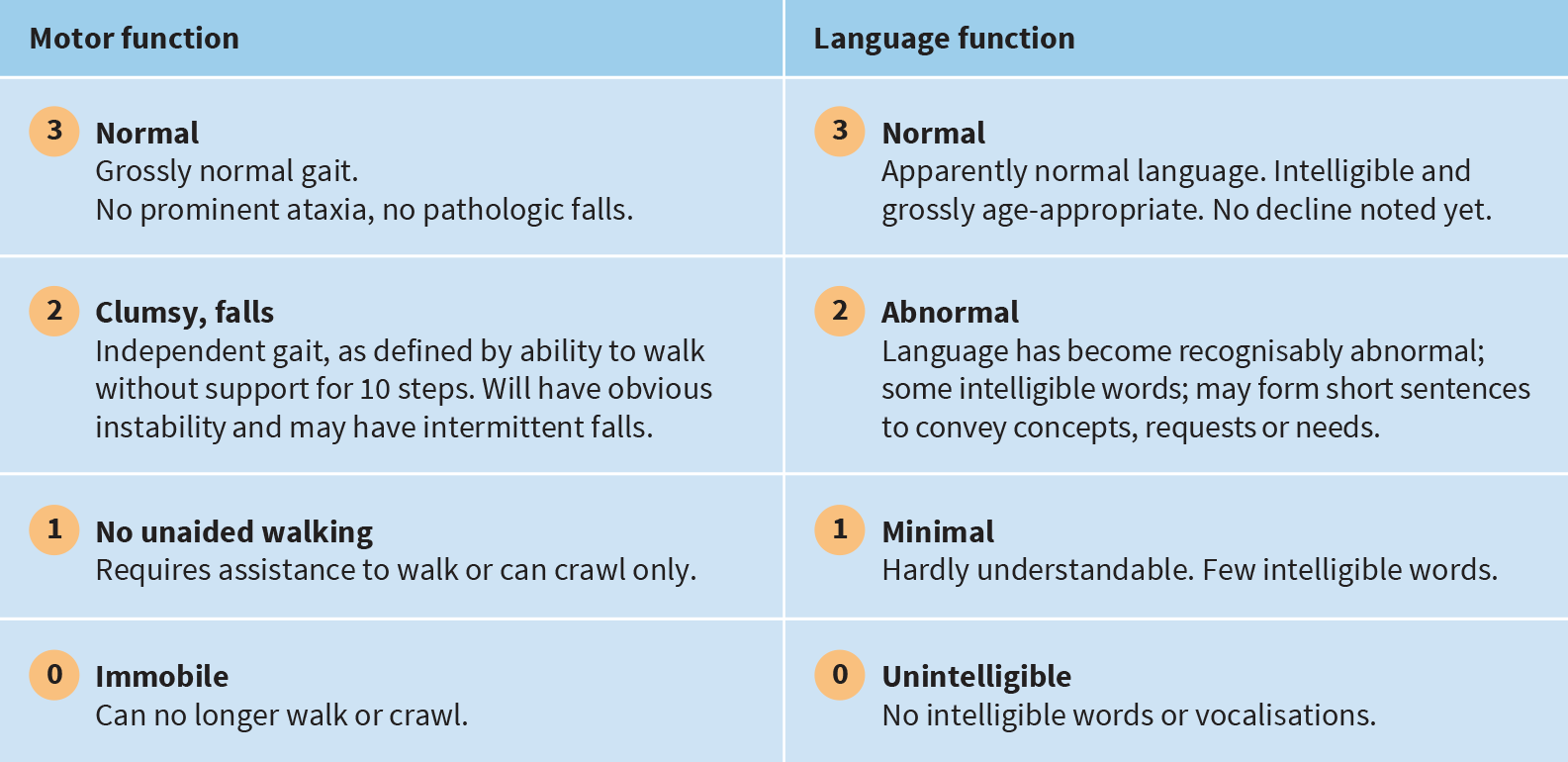
In addition to motor and language function assessments, there are also functional domains (each with a 3-point scale) to assess vision and the frequency of seizures.7
*In some children, normal motor and/or language development was never present. In such cases, the best performance achieved by the child was considered normal. When that performance level became recognisably worse, the child was rated a 2 for slightly abnormal motor function and/or language development.
Dr Alfried Kohlschuetter, MD
Professor of Paediatrics
Department of Paediatrics
University Medical Center Hamburg-Eppendorf
Hamburg, Germany
Professor of Paediatrics
Department of Paediatrics
University Medical Center Hamburg-Eppendorf
Hamburg, Germany
Hear how the CLN2 disease clinical rating scale was created to help physicians quantitatively track disease progression, and learn more about the important role carers play in providing disease progression insights as they track and monitor routine functions in their child with CLN2 disease.
Recognising CLN2 disease is a race against time
The majority of children with CLN2 disease experience a consistent loss of motor and language function, as measured by the CLN2 disease clinical rating scale8

Longitudinal data from 58 subjects with CLN2 disease in DEM-CHILD registry.
- Average age of first seizure: ~3 years
- Average age of CLN2 disease diagnosis: ~5 years
- Average loss per year: 2 points
- Average age of death: 10.1 years
With a rapid rate of progression, 2 years to diagnosis is too long.
References: 1. Schulz A, et al. NCL diseases — clinical perspectives. Biochim Biophys Acta. 2013;1832:1801 -1806. 2. Mole SE, Williams RE. Neuronal ceroid-lipofuscinoses. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReview® [internet]. Seattle, WA: University of Washington;1993 -2016. 3. Mole SE et al. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics 2005;6:107 -126. 4. Chang M et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom:Oxford University Press; 2011:80 -109. 5. Pérez-Poyato MS et al. Late Infantile Neuronal Ceroid Lipofuscinosis: Mutations in the CLN2 Gene and Clinical Course in Spanish Patients. J Child Neurol 2013;28:470 -478. 6. Worgall S et al. Treatment of Late Infantile Neuronal Ceroid Lipofuscinosis by CNS Administration of a Serotype 2 Adeno-Associated Virus Expressing CLN2 cDNA. Hum Gene Ther 2008;19:463 -474. 7. Steinfeld R et al. Late Infantile Neuronal Ceroid Lipofuscinosis: Quantitative Description of the Clinical Course in Patients With CLN2 Mutations. Am J Med Genet 2002;112:347 -354. 8. Nickel M et al. Disease characteristics and progression in patients with late-infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease: an observational cohort study. Lancet Child Adolesc Health 2018;2(8):582–590. 9. Fietz M et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): Expert recommendations for early detection and laboratory diagnosis. Mol Genet Metab. 2016;119:160–167. 10. Schulz A et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N Engl J Med. 2018;378:1898–907.
