 Русский
Русский
 Español
Español UK (English)
UK (English) Deutsch
Deutsch Italiano
Italiano Français
Français
Forget your password?
Патофизиология

Патофизиология
На фоне дефицита фермента TPP1 у детей с НЦЛ2 развивается нейродегенерация1
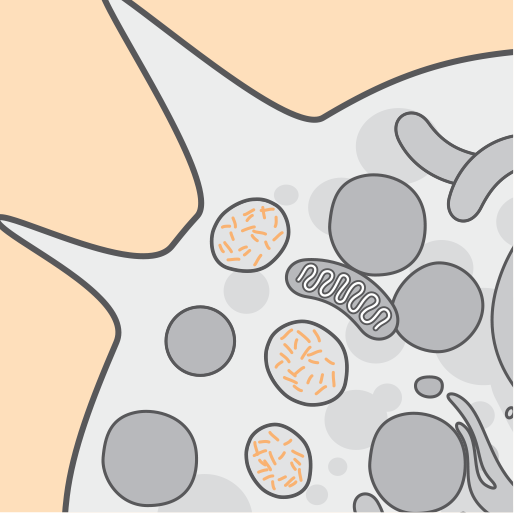
Мутации гена CLN2/TPP1 приводят к недостаточной активности фермента трипептидил пептидазы 1 (TPP1). TPP1 представляет собой протеазу, которая отщепляет n-концевой трипептид от субстрата в лизосомах1,2.
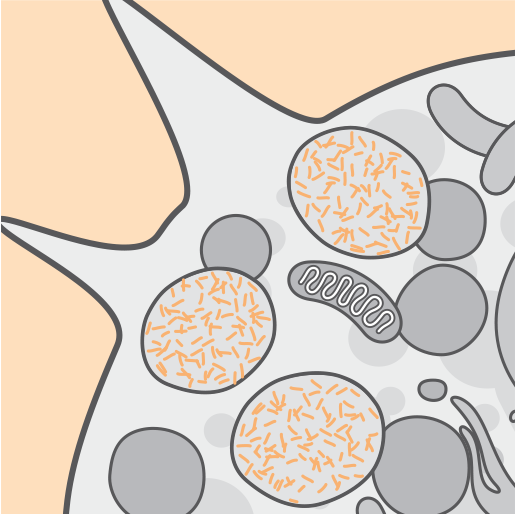
В результате отсутствия или снижения активности фермента TPP1 происходит накопление лизосомного автофлуоресцентного липидогенного пигмента (цероидного липофусцина)2,3.
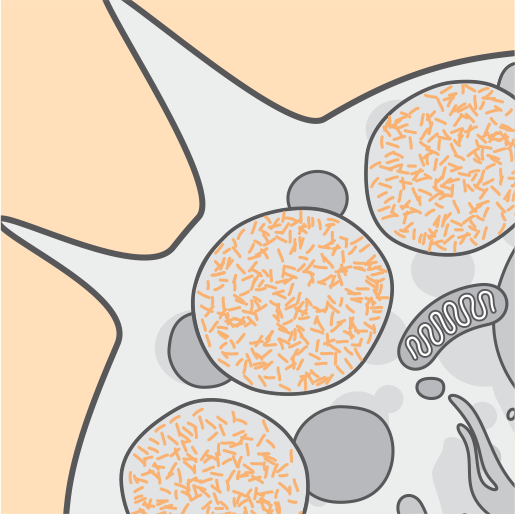
Со временем возникает дисфункция клеток, происходит их гибель и атрофия3,4.
Непрерывное накопление в лизосомах повреждает клетки, очевидными становятся разрушительные симптомы НЦЛ2.
Источники: 1. Schulz A et al. NCL diseases – clinical perspectives [Заболевания НЦЛ: клинические перспективы]. Biochimica et Biophysica Acta. 2013;1832:1801-1806. 2. Mole SE et al. Correlations between genotype, ultra structural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses [Корреляции между генотипом, ультраструктурной морфологией и клиническим фенотипом при нейрональном цероидном липофусцинозе]. Neurogenetics. 2005;6:107-126. 3. Chang M et al. CLN2. In Mole S, Williams R, Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease) [Нейрональный цероидный липофусциноз (болезнь Баттена)]. 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 4. Haltia M. The neuronal ceroid-lipofuscinoses: from past to present [Нейрональный цероидный липофусциноз: прошлое и настоящее]. Biochimica et Biophysica Acta. 2006;1762:850-856.
