 Türkçe
Türkçe
 Español
Español UK (English)
UK (English) Deutsch
Deutsch Italiano
Italiano Русский
Русский Français
Français
Forget your password?
Patofizyoloji

Patofizyoloji
TPP1 enzimi yetersizliği, CLN2 hastalığı olan çocuklarda nörodejenerasyonla sonuçlanır1
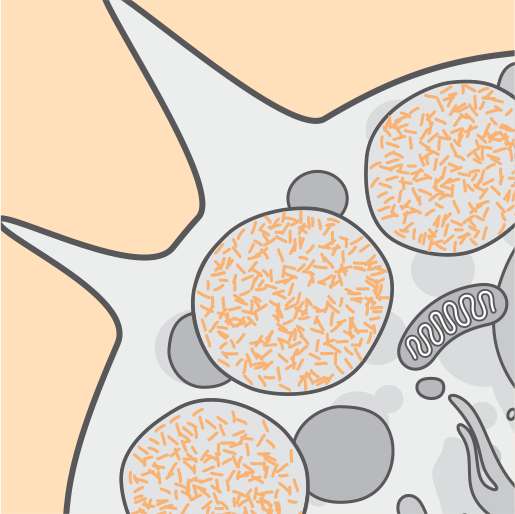
Mutations in the CLN2/TPP1 gene result in deficient activity of the tripeptidyl-peptidase 1 (TPP1) enzyme. TPP1 is a protease that cleaves N-terminal tripeptides from substrates in lysosomes.3-5
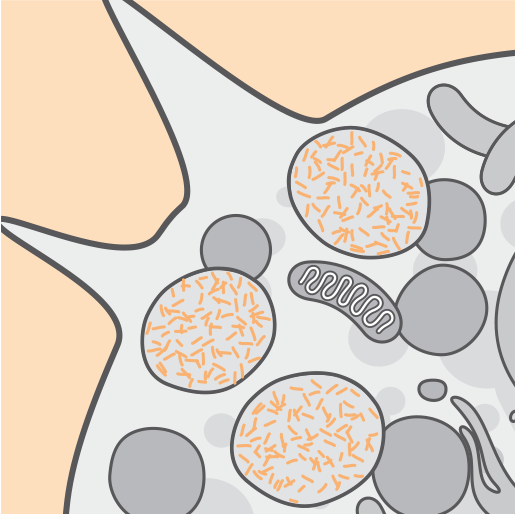
TPP1 enziminin yokluğu veya azalan aktivitesi, lizozomal otofloresan lipopigment (seroid lipofuksin) depo maddesinin birikmesiyle ilişkilidir.2,3
Zaman içinde hücre disfonksiyonu, hücre ölümü ve atrofi meydana gelir.3,4
Lizozomlarda sürekli olarak birikme olması hücrelere zarar verir ve CLN2 hastalığının yıkıcı semptomları fark edilebilir hale gelir.
Referanslar: 1. Schulz A et al. NCL diseases – clinical perspectives. Biochimica et Biophysica Acta. 2013;1832:1801-1806. 2. Mole SE et al. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126. 3. Chang M et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 4. Haltia M. The neuronal ceroid-lipofuscinoses: from past to present. Biochimica et Biophysica Acta. 2006;1762:850-856.
