 Italiano
Italiano
 Español
Español UK (English)
UK (English) Deutsch
Deutsch Türkçe
Türkçe Русский
Русский Français
Français
Forget your password?
Decorso naturale

Decorso naturale
La CLN2 è caratterizzata da un decorso terribilmente rapido: i sintomi e la perdita funzionale si accentuano con l'età
Cronologia dell'insorgenza dei sintomi e della perdita funzionale nella CLN2 in forma classica1-6

Le fasce di età rappresentate sono valori medi relativi al fenotipo tardo-infantile classico. I fenotipi atipici della CLN2 possono variare per età di insorgenza, rapidità di avanzamento e manifestazione della malattia.
Da 0 a 2 anni
- Si ritiene che l'accumulo lisosomiale dei lipopigmenti abbia inizio nel periodo prenatale, seppure lo sviluppo dei bambini appaia normale fino all'insorgenza dei sintomi2
- Nella maggior parte dei casi, il primo sintomo è rappresentato dall’esordio tardivo del linguaggio, come dimostrato da recenti studi clinici7
- In pochi casi, altri ritardi nello sviluppo, difficoltà motorie o atassia possono rappresentare un primo segno4,5
- Tali ritardi possono spesso manifestarsi insieme all’esordio tardivo del linguaggio
Da 2 a 4 anni
- Per quanto l’esordio tardivo del linguaggio si manifesti in una fase precedente, le crisi epilettiche (che compaiono intorno ai 3 anni) rappresentano il sintomo che induce più spesso i genitori a sospettare la presenza della CLN2 e a richiedere assistenza medica2,8
- È possibile osservare diversi tipi di crisi epilettiche, comprese le forme generalizzate tonico-cloniche, di assenza, miocloniche, atoniche, cloniche e toniche2,4
- Sebbene le crisi epilettiche siano per lo più non provocate, possono manifestarsi anche crisi convulsive febbrili4
- L’atassia insorge spesso durante questa fase della malattia5
Da 4 a 7 anni
- La sintomatologia si consolida con l'età ed è accompagnata da una drastica perdita di capacità precedentemente acquisite. Solitamente, in questa fascia di età, i bambini affetti da CLN2 presentano la seguente sintomatologia:1,2,8,9
- Regressione del linguaggio
- Crisi miocloniche, sia epilettiche che non epilettiche
- Grave riduzione della funzione motoria con eventuale perdita di movimenti volontari
- Disturbi motori, quali mioclono, spasticità, distonia e corea
- Deterioramento visivo fino alla cecità all'età di 6 o 7 anni
- Si assiste infine alla perdita completa del linguaggio e della capacità di deambulazione; i bambini iniziano a dipendere dall'uso della sedia a rotelle, rimangono allettati e devono affidarsi completamente alle cure del personale sanitario2
Da 8 a 12 anni
- La CLN2 è caratterizzata da morte prematura e la maggior parte dei bambini riesce raramente a sopravvivere oltre la prima fase dell'adolescenza1
La cecità costituisce un primo sintomo in altre NCL, ma si verifica in una fase successiva della CLN2.6,9

Dott. Paul Gissen, MBChB, PhD, FRCPCH
University College London (UCL)
Ospedale pediatrico Great Ormond Street
NHS Foundation Trust
University College London (UCL)
Ospedale pediatrico Great Ormond Street
NHS Foundation Trust
Panoramica della prevedibilità della CLN2 e delle fasi di sviluppo dei sintomi lungo l’intero arco di vita dei bambini colpiti.
La scala di valutazione clinica della CLN2 è un modo efficace di monitorare la progressione della malattia in 2 principali aree funzionali: funzione motoria e linguaggio
Il sistema di punteggio standardizzato può essere utilizzato per valutare in modo quantitativo la progressione della malattia in sede di diagnosi e monitorare l’evoluzione della perdita funzionale nel tempo6- A ciascuna area funzionale viene assegnato un punteggio su una scala da 3 (sostanzialmente normale) a 0 (profondamente compromessa)6
- Il punteggio massimo possibile nella valutazione della funzione motoria e del linguaggio è 67
Le prestazioni funzionali vengono valutate come segue:6
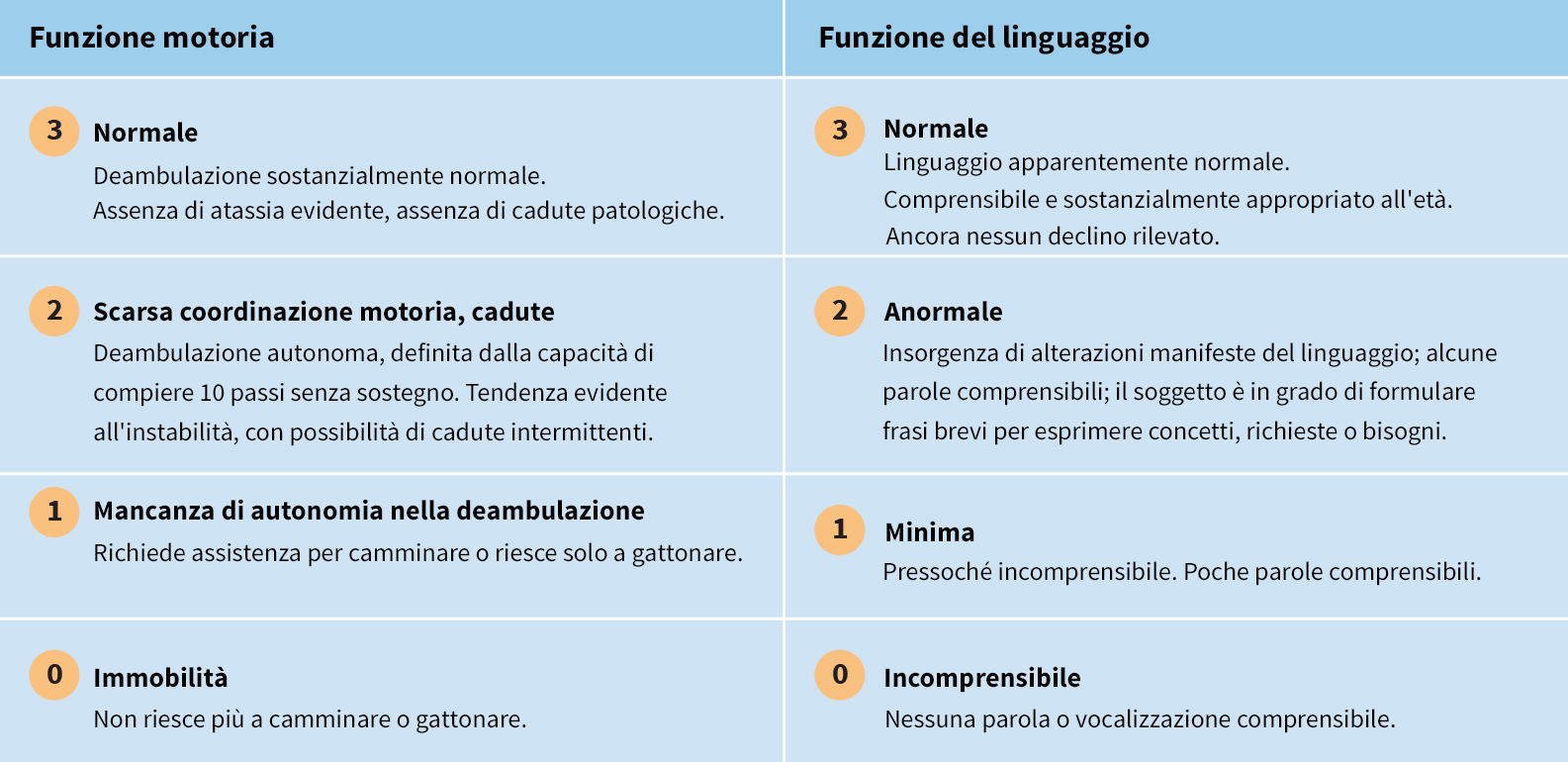
Oltre alla valutazione della funzione motoria e del linguaggio, sono previsti anche domini funzionali (ognuno con una scala a 3 punti) atti a valutare la vista e la frequenza delle crisi epilettiche.6
* In alcuni bambini, non è mai stato riscontrato un normale sviluppo della funzione motoria e/o del linguaggio. In questi casi, sono state considerate normali le prestazioni migliori ottenute dal bambino. In seguito al notevole peggioramento del livello delle prestazioni, al bambino è stato assegnato un punteggio pari a 2, in presenza di una lieve riduzione della funzione motoria e/o dello sviluppo del linguaggio.
Dott. Alfried Kohlschuetter, MD
Professore di Pediatria
Dipartimento pediatrico
Centro medico universitario di Amburgo-Eppendorf
Amburgo, Germania
Professore di Pediatria
Dipartimento pediatrico
Centro medico universitario di Amburgo-Eppendorf
Amburgo, Germania
Ascoltiamo in che modo è stata creata la scala di valutazione clinica della CLN2 per supportare i medici nel monitoraggio quantitativo della progressione della malattia. La conoscenza diretta della progressione della malattia, derivante dall'osservazione delle normali funzioni del bambino affetto da CLN2, permette di approfondire l'importanza del ruolo di divulgazione della stessa svolto dal personale di assistenza.
Il riconoscimento della CLN2 è una corsa contro il tempo
La maggior parte dei bambini affetti da CLN2 manifesta una perdita costante della funzione motoria e del linguaggio, misurata tramite la scala di valutazione clinica della CLN27

Dati longitudinali relativi a 58 soggetti affetti da CLN2 nel registro DEM-CHILD.
- Età media della prima crisi epilettica: ~3 anni
- Età media alla diagnosi della CLN2: ~5 anni
- Perdita media annua: 2 punti
- Età media di decesso: 10,1 anni
Con un tasso di progressione elevato, un tempo di attesa diagnostica di 2 anni è eccessivo.
Bibliografia: 1. Mole SE et al. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126. 2. Chang M et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 3. Schulz A et al. NCL diseases – clinical perspectives. Biochimica et Biophysica Acta. 2013;1832:1801–1806. 4. Pérez-Poyato et al. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. J Child Neurol. 2013;28:470-478. 5. Worgall S et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum Gene Ther. 2008;19:463-474. 6. Steinfeld R et al. Late infantile neuronal ceroid lipofuscinosis: quantitative description of the clinical course in patients with CLN2 mutations. Am J Med Genet. 2002;112:347-354. 7. Nickel M et al. Natural history of CLN2 disease: quantitative assessment of disease characteristics and rate of progression. Poster session presented at: The 12th Annual WORLD Symposium; February – March 2016; San Diego, CA. 8. Schulz A et al. Neuronal ceroid lipofuscinosis-2 (CLN2) disorder, a type of Batten disease caused by TPP1 enzyme deficiency: current knowledge of the natural history from international experts. Poster session presented at: The Society for the Study of Inborn Errors of Metabolism (SSIEM) Annual Symposium; September 2015; Lyon, France. 9. Mole SE and Williams RE. Neuronal ceroid-lipofuscinoses. 2001 Oct 10 [Updated 2013 Aug 1]. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews® [internet]. Seattle, WA: University of Washington; 1993-2016.
