 Italiano
Italiano
 Español
Español UK (English)
UK (English) Deutsch
Deutsch Türkçe
Türkçe Русский
Русский Français
Français
Forget your password?
Necessità di una diagnosi precoce

Necessità di una diagnosi precoce
I segni e i sintomi della CLN2 non sono limitati all'epilessia
I ritardi nella diagnosi della CLN2 sono comuni
- L'età media alla diagnosi è di 5 anni, corrispondente a una fase avanzata del declino neurodegenerativo della CLN21
- Il ritardo nella diagnosi della CLN2 può dipendere da vari fattori1:
- Sintomi iniziali non specifici
- Bassa consapevolezza della malattia
- La diagnosi precoce consente di ricorrere a cure cliniche e assistenza sociale
specifiche per la CLN2 che possono produrre effetti positivi sui risultati clinici e sulla qualità della vita del bambino e dei familiari1,2 - La diagnosi precoce della CLN2 è fondamentale ai fini della consapevolezza dei genitori di essere portatori sani e rafforza l'importanza di una pianificazione familiare3
- I ritardi nella diagnosi possono essere accompagnati da valutazioni approfondite e diagnosi errate prima che venga raggiunta una diagnosi definitiva2,4
La diagnosi della CLN2 viene ritardata in media di 2 anni dalla prima crisi epilettica osservata all'età di 3 anni.
Spesso la CLN2 non viene inizialmente diagnosticata in maniera corretta, con conseguente ritardo in termini di accuratezza
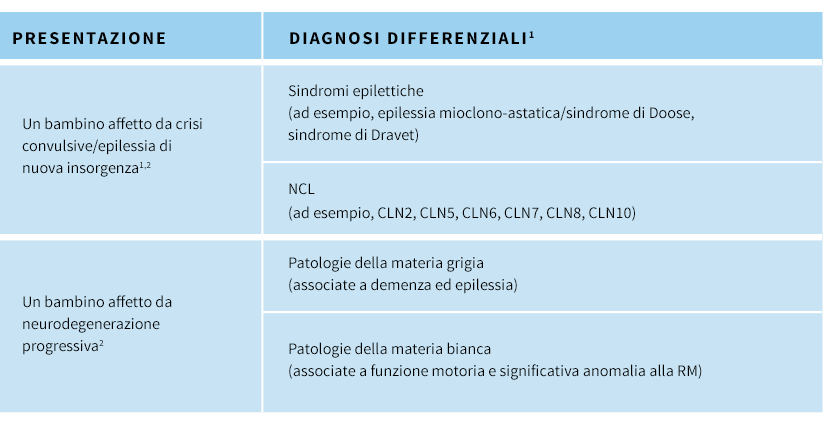
La diagnosi differenziale della CLN2 varia a seconda della fase di progressione della malattia.Fase iniziale nella progressione della CLN2:3,5
- Dal momento che le crisi epilettiche e il mioclono costituiscono i sintomi più evidenti, le sindromi in cui il mioclono è comune (ad esempio, l’epilessia focale, le sindromi di epilessia mioclonica/epilessie mioclono-astatiche quali la sindrome di Doose, la sindrome di Dravet, la sindrome di Lennox-Gastaut o altre sindromi epilettiche) destano frequentemente sospetti
Fase tardiva nella progressione della CLN2:3
- Con la progressione della malattia e la maggiore evidenza di regressione psicomotoria e perdita funzionale, possono essere sospettati altri disturbi cerebrali pediatrici progressivi (ad esempio, infiammazioni/infezioni, tumori, disturbi mitocondriali e altre malattie da accumulo lisosomiale, compresi altri tipi di NCL)
Bibliografia: 1. Fietz M et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): Expert recommendations for early detection and laboratory diagnosis. Mol Genet Metab. 2016; 119:160–167 2. Williams RE et al. Expert opinion on the management of CLN2 disease. Poster session presented at: The 12th Annual WORLD Symposium; February – March 2016; San Diego, CA. 3. Chang M et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 4. Pérez-Poyato MS et al. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. J Child Neurol. 2013;28:470-478. 5. Mole SE, Williams RE, and Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126.
