 Italiano
Italiano
 Español
Español UK (English)
UK (English) Deutsch
Deutsch Türkçe
Türkçe Русский
Русский Français
Français
Forget your password?
Fisiopatologia

Fisiopatologia
Il deficit dell'attività enzimatica della TPP1 determina la neurodegenerazione nei bambini affetti da CLN21
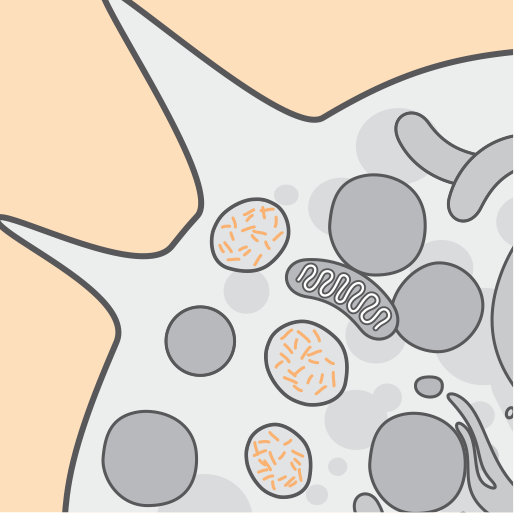
Le mutazioni nel gene CLN2/TPP1 determinano un deficit dell'attività dell'enzima tripeptidil-peptidasi 1 (TPP1). La TPP1 è una proteasi che scinde i tripeptidi N-terminali dai substrati dei lisosomi.1,2
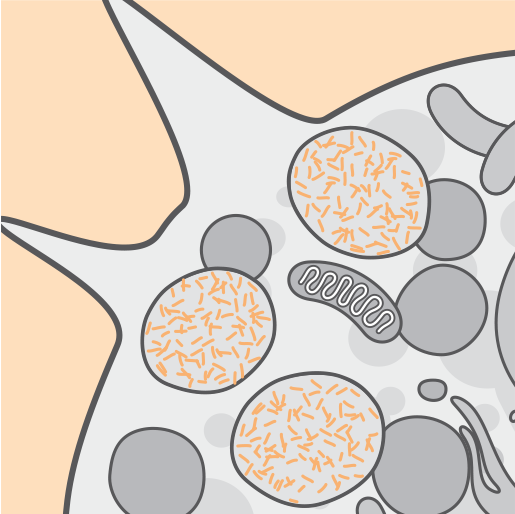
L'assenza o la ridotta attività dell'enzima TPP1 è associata a un accumulo lisosomiale di lipopigmenti autofluorescenti (lipofuscina ceroide).2,3
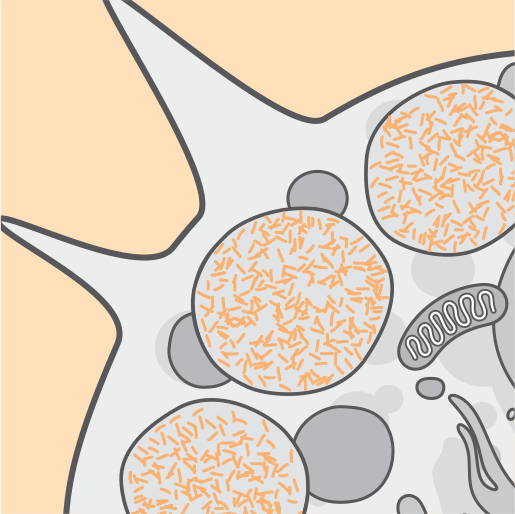
Nel corso del tempo si verificano disfunzioni cellulari, morte cellulare e atrofia.3,4
Il continuo accumulo nei lisosomi danneggia le cellule e i sintomi devastanti della CLN2 diventano evidenti.
Bibliografia: 1. Schulz A et al. NCL diseases – clinical perspectives. Biochimica et Biophysica Acta. 2013;1832:1801-1806. 2. Mole SE et al. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126. 3. Chang M et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 4. Haltia M. The neuronal ceroid-lipofuscinoses: from past to present. Biochimica et Biophysica Acta. 2006;1762:850-856.
