Diese Seite ist nur für medizinisches Fachpersonal bestimmt. Bitte click here wenn Sie keine medizinische Fachkraft sind.
 Deutsch
Deutsch
 Deutsch
Deutsch Español
Español UK (English)
UK (English) Türkçe
Türkçe Italiano
Italiano Русский
Русский Français
Français
Forget your password?
Natürlicher Krankheitsverlauf

Natürlicher Krankheitsverlauf
Die CLN2-Krankheit weist einen verheerend schnellen Verlauf auf – Symptome und Funktionsverlust verschlimmern sich mit zunehmendem Alter
Zeitstrahl des Auftretens von Symptomen und Funktionsverlusts bei der klassischen CLN2-Krankheit1-6

Die dargestellten Altersbereiche stellen Durchschnittswerte für den klassisch spät-infantilen Phänotyp dar. Atypische Phänotypen der CLN2-Krankheit können sich hinsichtlich Alter beim Ausbruch der Krankheit, Progressionsrate und Manifestation der Erkrankung unterscheiden.
0 bis 2 Jahre
- Anreicherung von Lipopigmenten in den Lysosomen beginnt wahrscheinlich bereits in der Pränatalzeit, jedoch entwickeln sich die Kinder bis zum Einsetzen von Symptomen scheinbar normal2
- Wie neueste klinische Befunde belegen, besteht in den meisten Fällen das erste Symptom in einer früh auftretenden Sprachentwicklungsverzögerung7
- In einigen wenigen Fällen können andere Entwicklungsverzögerungen, Bewegungsstörungen oder Ataxie erste
Anzeichen darstellen4,5- Diese können auch häufig gemeinsam mit einer Sprachentwicklungsverzögerung auftreten
2 bis 4 Jahre
- Selbst wenn eine Sprachentwicklungsverzögerung früher auftritt, sind Krampfanfälle (treten meistens ab 3 Jahren auf) im Allgemeinen das Symptom, aufgrund dessen Eltern einen Arzt aufsuchen, um ihr Kind auf die CLN2-Krankheit hin untersuchen zu lassen2,8
- Verschieden Arten von Krampfanfällen können auftreten, einschließlich generalisierte tonisch-klonische, myoklonische, atonische, klonische und tonische Anfälle sowie Absence-Anfälle2,4
- Auch wenn Krampfanfälle meist unprovoziert auftreten, sind Fieberkrämpfe möglich4
- In diesem Krankheitsstadium kommt es häufig zum Einsetzen von Ataxie5
4 bis 7 Jahre
- Symptome verschlechtern sich mit zunehmendem Alter, und es findet ein spürbarer Verlust zuvor erworbener Fertigkeiten statt. In diesem Altersbereich treten bei an CLN2 erkrankten Kindern normalerweise folgende Symptome auf:1,2,8,9
- Rückgang der Sprachfertigkeit
- Myoklonische Krampfanfälle sowohl epileptischer als auch nicht epileptischer Natur
- Schwere Beeinträchtigung der motorischen Funktion, was schließlich zum Verlust der willkürlichen Bewegungen führt
- Bewegungsstörungen wie Myoklonie, Spastik, Dystonie und Chorea
- Verschlechterung der Sehkraft, was im Alter von 6 oder 7 Jahren zur Erblindung führt
- Letztendlich kommt es zu einem vollständigen Verlust von Sprache und Gehvermögen, und die Kinder sind auf einen Rollstuhl angewiesen, werden bettlägerig und benötigen eine Pflegeperson2
8 bis 12 Jahre
- Die CLN2-Krankheit ist geprägt von vorzeitigem Tod und die Mehrheit der Kinder erreicht meist nur die frühe Pubertät1
Während Erblindung ein früh auftretendes Symptom bei anderen NCL ist, tritt sie bei der CLN2-Krankheit erst im späteren Stadium auf.6,9

Dr. Paul Gissen, MBChB, PhD, FRCPCH
University College London
Kinderklinik Great Ormond Street
NHS Foundation Trust
University College London
Kinderklinik Great Ormond Street
NHS Foundation Trust
Ein Überblick über die vorhersagbare Natur der CLN2-Krankheit und zu welchem Zeitpunkt im Leben der betroffenen Kinder die Symptome auftreten.
Die Beurteilungsskala der CLN2-Krankheit stellt eine effiziente Möglichkeit dar, die Krankheitsprogression in 2 wichtigen Funktionsbereichen zu beurteilen: motorische und sprachliche Fähigkeit
Das standardisierte Bewertungssystem kann während der Diagnose zur quantitativen Beurteilung der Krankheitsprogression sowie zur genaueren Beurteilung des Funktionsverlusts im Laufe der Zeit eingesetzt werden6
- Jeder Funktionsbereich wird auf einer Skala von 3 (weitgehend normal) bis 0 (äußerst stark beeinträchtigt) bewertet6
- Der höchstmögliche Wert, der bei der Beurteilung der motorischen und sprachlichen Funktionen zu erreichen ist,
beträgt 67
Bewertungsskala zur funktionsbezogenen Leistungsfähigkeit:6
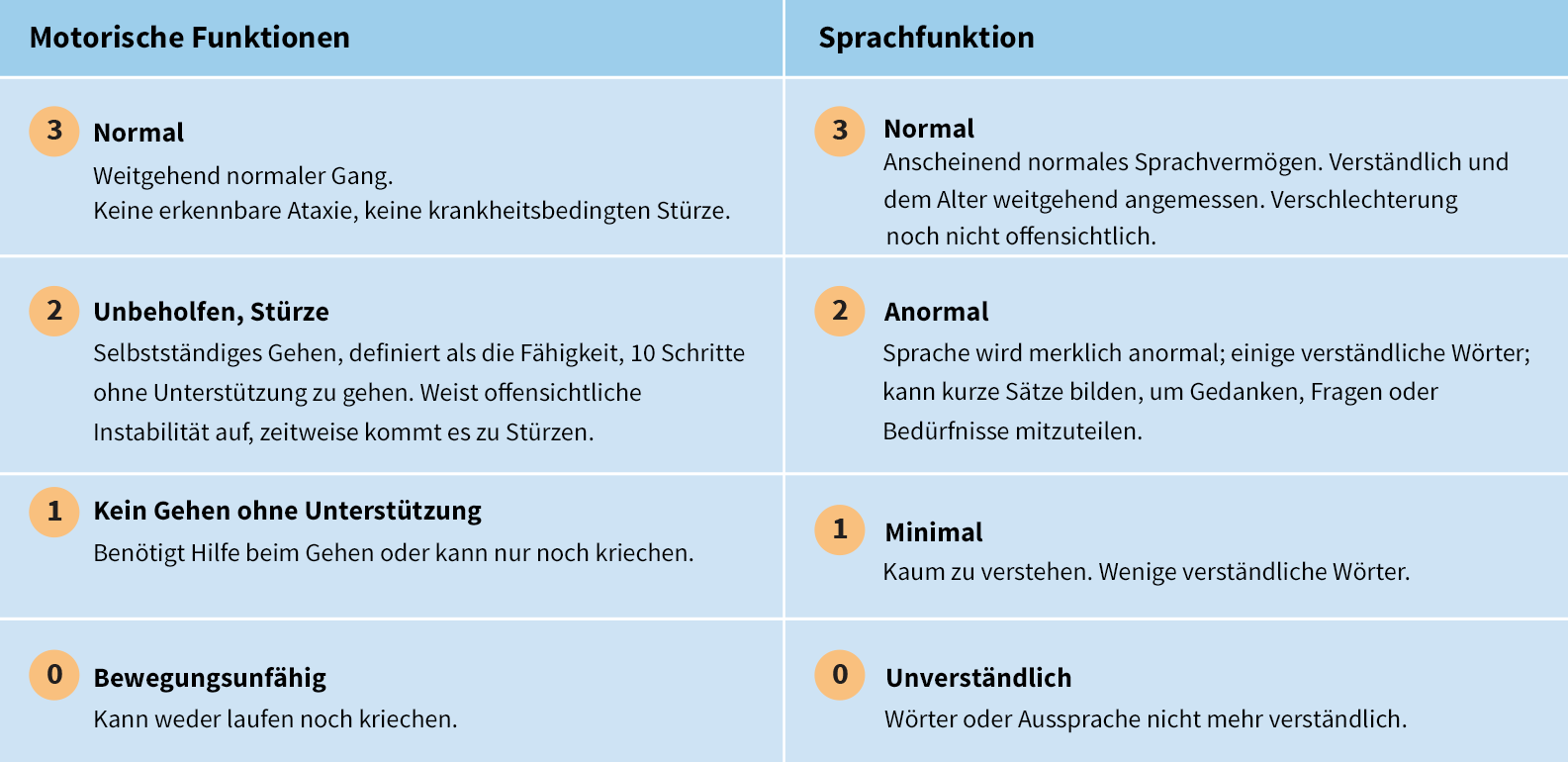
In addition to motor and language function assessments, there are also functional domains (each with a 3-point scale) to assess vision and the frequency of seizures.7
*In some children, normal motor and/or language development was never present. In such cases, the best performance achieved by the child was considered normal. When that performance level became recognisably worse, the child was rated a 2 for slightly abnormal motor function and/or language development.
Dr. med. Alfried Kohlschütter
Professor für Kinder- und Jugendmedizin
Institut für Kinder- und Jugendmedizin
Universitätsklinikum Hamburg-Eppendorf
Hamburg, Deutschland
Professor für Kinder- und Jugendmedizin
Institut für Kinder- und Jugendmedizin
Universitätsklinikum Hamburg-Eppendorf
Hamburg, Deutschland
Hören Sie, wie die klinische Beurteilungsskala der CLN2-Krankheit erstellt wurde, um Ärzten dabei zu helfen, die Krankheitsprogression quantitativ nachzuvollziehen und erfahren Sie mehr darüber, welche wichtige Rolle Bezugspersonen spielen bei der Erlangung von Einblicken in die Krankheitsprogression, indem sie die grundlegenden Funktionen bei ihrem an CLN2 erkrankten Kind verfolgen und überwachen.
CLN2-Krankheit: Ein Wettlauf gegen die Zeit
Bei der Mehrzahl der an CLN2 erkrankten Kinder kommt es zu einem gleichmäßigen Verlust der motorischen und sprachlichen Funktion, gemessen mit der Beurteilungsskala der CLN2-Krankheit7

Longitudinale Daten von 58 Patienten mit CLN2-Krankheit aus dem DEM-CHILD-Register. KI = Konfidenzintervall.
- Durchschnittsalter bei erstem Anfall: ~3 Jahre
- Durchschnittsalter bis Diagnose der CLN2-Krankheit: ~5 Jahre
- Durchschnittlicher Verlust pro Jahr: 2 Punkte
- Durchschnittsalter bei Tod: 10,1 Jahre
Aufgrund der raschen Progressionsrate sind 2 Jahre bis zur Diagnose zu lang.
Literaturangaben: 1. Mole SE, Williams RE, and Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126. 2. Chang M, Cooper JD, Davidson BL, et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 3. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases – clinical perspectives. Biochimica et Biophysica Acta. 2013;1832:1801–1806. 4. Pérez-Poyato MS, Marfa MP, Abizanda IF, et al. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. J Child Neurol. 2013;28:470-478. 5. Worgall S, Sondhi D, Hackett NR, et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum Gene Ther. 2008;19:463-474. 6. Steinfeld R, Heim P, von Gregory H, et al. Late infantile neuronal ceroid lipofuscinosis: quantitative description of the clinical course in patients with CLN2 mutations. Am J Med Genet. 2002;112:347-354. 7. Nickel M, Jacoby D, Lezius S, et al. Natural history of CLN2 disease: quantitative assessment of disease characteristics and rate of progression. Poster session presented at: The 12th Annual WORLD Symposium; February – March 2016; San Diego, CA. 8. Schulz A, Cohen-Pfeffer JL, Crystal R, et al. Neuronal ceroid lipofuscinosis-2 (CLN2) disorder, a type of Batten disease caused by TPP1 enzyme deficiency: current knowledge of the natural history from international experts. Poster session presented at: The Society for the Study of Inborn Errors of Metabolism (SSIEM) Annual Symposium; September 2015; Lyon, France. 9. Mole SE, Williams RE. Neuronal ceroid-lipofuscinoses. 2001 Oct 10 [Updated 2013 Aug 1]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [internet]. Seattle, WA: University of Washington; 1993-2016.
