Diese Seite ist nur für medizinisches Fachpersonal bestimmt. Bitte click here wenn Sie keine medizinische Fachkraft sind.
 Deutsch
Deutsch
 Deutsch
Deutsch Español
Español UK (English)
UK (English) Türkçe
Türkçe Italiano
Italiano Русский
Русский Français
Français
Forget your password?
Notwendigkeit einer frühen Diagnose

Notwendigkeit einer frühen Diagnose
Bei der CLN2-Krankheit gibt es noch andere Anzeichen und Symptome als bei einer Epilepsie
Dennoch wird bei Kindern häufig eine Epilepsie diagnostiziert, ohne die Möglichkeit einer CLN2 in Betracht zu ziehen
- Durchschnittsalter bei Diagnose liegt bei 5 Jahren – zu dieser Zeit ist der neurodegenerative Verfall durch die CLN2-Krankheit schon weit vorangeschritten1
- Gründe für eine verzögerte Diagnose der CLN2-Krankheit:1
- Initiale Symptome sind nichtspezifisch
- Progression der CLN2-Krankheit kann mit Nebenwirkungen der Antiepileptika verwechselt werden
- Durch eine frühe Diagnose ist der Zugang zu CLN2-spezifischer klinischer Versorgung und sozialer Unterstützung möglich, was die Behandlungserfolge und die Lebensqualität des Kindes und seiner Familie positiv beeinflussen kann1,2
- Eine frühzeitige Diagnose der CLN2-Krankheit ist entscheidend, damit Eltern sich bewusst werden, dass sie Träger des Gens sind, und untermauert die Bedeutung der Familienplanung3
- Bei einer verzögerten Diagnose kann es zu umfangreichen Untersuchungen und Fehldiagnosen kommen, bevor eine definitive Diagnose bestätigt wird2,4
Bis es nach ersten beobachteten Krampfanfällen im Alter von 3 Jahren zur Diagnose der CLN2-Krankheit kommt, vergehen im Durchschnitt 2 Jahre.
Bei einer vorliegenden CLN2-Krankheit kommt es zu Beginn häufig zu Fehldiagnosen, was eine korrekte Diagnose verzögert
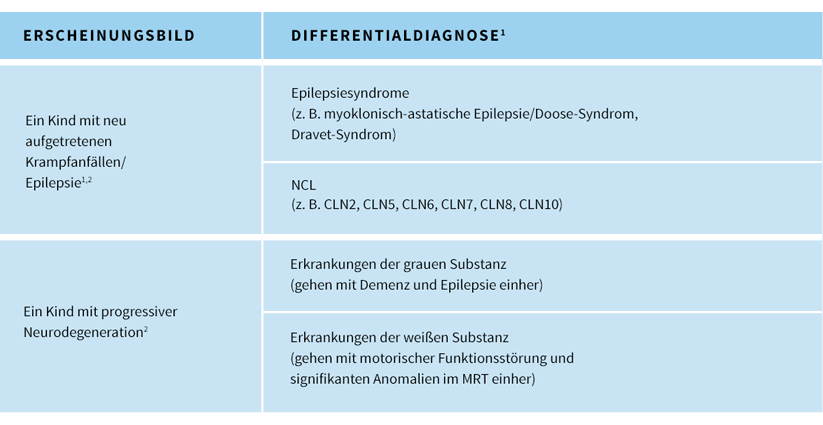
Die Differentialdiagnose bei der CLN2-Krankheit variiert in Abhängigkeit vom Stadium der Krankheitsprogression.Während des Frühstadiums der CLN2-Krankheit:3,5
- Da Krampfanfälle und Myoklonien die auffälligsten Symptome darstellen, werden oftmals Krankheitsbilder vermutet, bei denen Myoklonien häufig auftreten (z. B. fokale Epilepsie, myoklonische Epilepsiesyndrome/myoklonisch-astatische Epilepsien wie Doose-Syndrom, Dravet-Syndrom, Lennox-Gastaut-Syndrom oder andere Epilepsiesyndrome)
Während des Spätstadiums der CLN2-Krankheit:3
- Während die Krankheit weiter voranschreitet und psychomotorische Regression und Funktionsverlust immer offensichtlicher werden, können andere progressiv verlaufende pädiatrische Gehirnerkrankungen als mögliche Ursache im Verdacht stehen (z. B. Entzündungen/Infektionen, Tumoren, mitochondriale Erkrankungen sowie andere lysosomale Speicherkrankheiten, einschließlich andere NCL-Typen)
Literaturangaben: 1. Fietz M, Giugliani R, AlSayed M, et al. Expert recommendations for the laboratory diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): diagnostic algorithm and best practice guidelines for a timely diagnosis. Poster session presented at: The 12th Annual WORLD Symposium; February – March 2016; San Diego, CA. 2. Williams RE, Adams HR, Blohm M, et al. Expert opinion on the management of CLN2 disease. Poster session presented at: The 12th Annual WORLD Symposium; February – March 2016; San Diego, CA. 3. Chang M, Cooper JD, Davidson BL, et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 4. Pérez-Poyato MS, Marfa MP, Abizanda IF, et al. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. J Child Neurol. 2013;28:470-478. 5. Mole SE, Williams RE, and Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126.
