Diese Seite ist nur für medizinisches Fachpersonal bestimmt. Bitte click here wenn Sie keine medizinische Fachkraft sind.
 Deutsch
Deutsch
 Deutsch
Deutsch Español
Español UK (English)
UK (English) Türkçe
Türkçe Italiano
Italiano Русский
Русский Français
Français
Forget your password?
NCL-Erkrankungen

NCL-Erkrankungen
Die CLN2-Krankheit ist eine der häufigsten Formen der NCL und ist auch unter dem Begriff Morbus Batten bekannt1,2
Es gibt 14 bekannte Formen der neuronalen Ceroid-Lipofuszinose (NCL)3
NCL besteht aus einer Gruppe von Erkrankungen, die maßgeblich für das Entstehen von Demenz bei Kindern und Jugendlichen verantwortlich sind.3
Die Nomenklatur der NCL-Erkrankungen wurde kürzlich überarbeitet und neu gegliedert und berücksichtigt nun die mutierten Gene und klinischen Anzeichen4
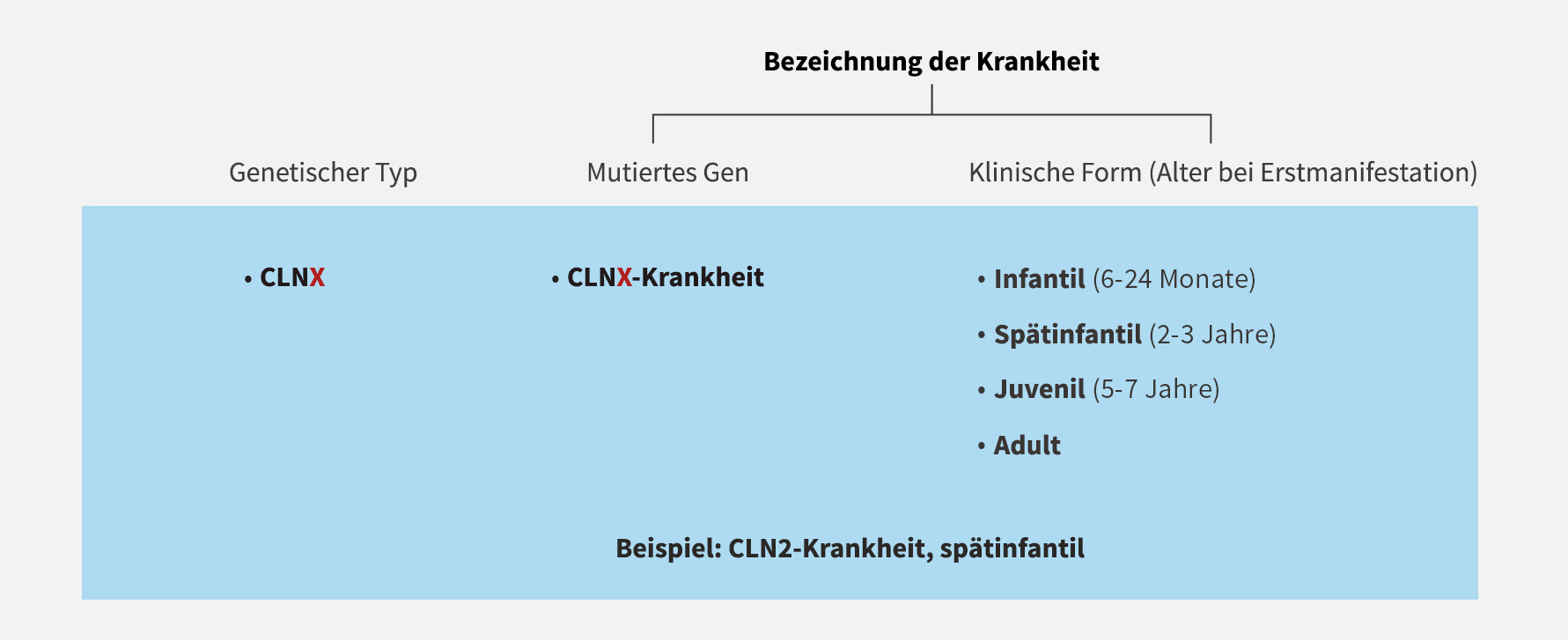
Klassifikation und Merkmale der NCL4-6
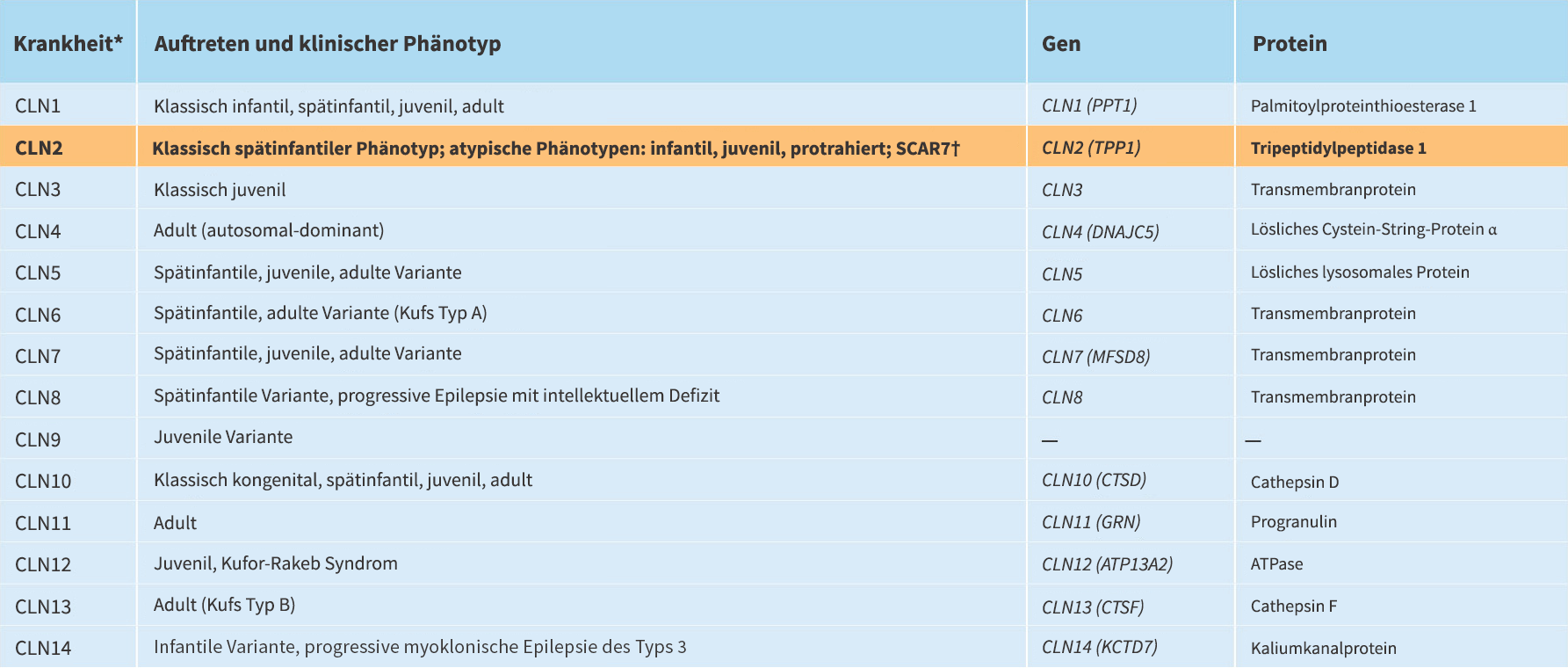
*Bekannte NCL-Erkrankungen. Weitere NCL-Gene müssen noch identifiziert werden.
†SCAR7 wird durch einen TPP1-Mangel verursacht und scheint eine schwächere Form von CLN2 zu sein.
CLN2 und CLN3 sind die weltweit am häufigsten auftretenden Formen von NCL.1
NCL-Erkrankungen gleichen sich in einer Vielzahl von ätiologischen und klinischen Merkmalen3
- Schwere und fortschreitende Neurodegeneration1
- Belastende Symptomtrias – Krampfanfälle, Demenz und Erblindung3
- Neuronale und retinale Akkumulation von Ceroid-Lipofuszin7
- NCLs weisen eindeutige Inklusionskörper auf, darunter solche mit granularen, kurvenförmigen und fingerabdruckähnlichen Profilen8
- Bei CLN2 ist die deutlich kurvenförmige Struktur der Ceroid-Lipofuszine unter dem Mikroskop erkennbar8
- Klassische und atypische Erscheinungsbilder, wobei das unterschiedliche Alter beim Ausbruch der Krankheit, Symptome und Krankheitsverläufe von Typ und Phänotyp abhängen3

Ultrastrukturelle Erscheinungsbilder verschiedener NCL-Formen, Haltia (2003)8
Literaturangaben:
1. Mole SE, Williams RE. Neuronal ceroid-lipofuscinoses. 2001 Oct 10 [Updated 2013 Aug 1]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [internet]. Seattle, WA: University of Washington; 1993-2016. 2. Haltia M. The neuronal ceroid-lipofuscinoses: from past to present. Biochimica et Biophysica Acta. 2006;1762:850-856. 3. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases – clinical perspectives. Biochimica et Biophysica Acta. 2013;1832:1801-1806. 4. Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. 2012;79:183-191. 5. Kousi M, Lehesjoki A-E, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat. 2012;33:42-63. 6. Fietz M, AlSayed M, Burke D, et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): Expert recommendations for early detection and laboratory diagnosis. [Epub ahead of print July 25, 2016]. Mol Genet Metab. doi: 10.1016/j.ymgme.2016.07.011. 7. Mole SE, Williams RE, and Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126.
