 Español
Español
 Español
Español UK (English)
UK (English) Deutsch
Deutsch Italiano
Italiano Türkçe
Türkçe Русский
Русский Français
Français
Forget your password?
Evolución natural de la enfermedad

Evolución natural de la enfermedad
En la enfermedad CLN2, los niños experimentan una dramática pérdida de función a medida que los síntomas progresan con la edad1
La neurodegeneración en niños con enfermedad CLN2 se asocia con deficiencia de la enzima TPP111,2

Se representan los rangos de edad promedio para el fenotipo tardío-infantil clásico. Los fenotipos atípicos de la CLN2 pueden diferir en la edad de inicio, tasa de progresión y manifestación de la enfermedad.
De 0 a 2 años
- El acúmulo lisosómico de lipopigmentos probablemente empieza en el periodo prenatal, y los niños parecen presentar un desarrollo normal hasta el inicio de los síntomas4
- En la mayoría de casos, el primer síntoma es un retraso del lenguaje temprano, tal como demuestran los hallazgos clínicos recientes8
- En una minoría de casos, el primer signo pueden ser los retrasos en el desarrollo, dificultades motoras o ataxia4,5
- A menudo pueden presentarse junto a retrasos en la adquisición del lenguaje5,8,9
De 2 a 4 años
- Aunque el retraso del lenguaje puede aparecer de forma temprana, las convulsiones (que aparecen sobre los 3 años de edad) son el síntoma que con más frecuencia impulsa a los padres a buscar atención médica en la CLN24,8
- Pueden observarse múltiples tipos de convulsiones, incluyendo convulsiones tonicoclónicas generalizadas, crisis de ausencia, mioclónicas, atónicas, clónicas y tónicas4,5
- Aunque la mayoría de las veces las convulsiones no son provocadas, también pueden presentarse convulsiones febriles5
- El inicio de la ataxia suele presentarse durante esta etapa de la enfermedad6
De 4 a 7 años
- Los síntomas se agravan con la edad y aparece una pérdida notable de las habilidades adquiridas. Durante este intervalo de edad, el niño con CLN2 presenta normalmente:2–4,10
- Regresión del lenguaje
- Convulsiones mioclónicas, tanto epilépticas como no epilépticas
- Deterioro grave de la función motora, con pérdida de los movimientos voluntarios
- Trastornos del movimiento tales como mioclonía, espasticidad, distonía y corea
- Deterioro visual que conduce a la ceguera a la edad de 6 o 7 años
- Finalmente, aparece una pérdida total del lenguaje y de la deambulación. Algunos niños quedan confinados a sillas de ruedas o encamados y totalmente dependientes de los cuidadores4
De 8 a 12 años
- La CLN2 se caracteriza por una mortalidad prematura. La mayoría de los niños raramente sobrevive más allá del inicio de la adolescencia3
La ceguera es un síntoma temprano en otras CLN, pero en la CLN2 se presenta en un estado tardío de la enfermedad.2,7

Dr. Paul Gissen, MBChB, PhD, FRCPCH
University College London
Hospital infantil Great Ormond Street
NHS Foundation Trust
University College London
Hospital infantil Great Ormond Street
NHS Foundation Trust
Una visión general de la naturaleza predecible de la CLN2 y del desarrollo de los síntomas durante la vida de los niños afectados.
La escala de valoración clínica de la CLN2 es una forma eficiente de evaluar la progresión de la enfermedad en dos áreas funcionales importantes: la función motora y el lenguaje
El sistema de puntuación estandarizado puede utilizarse para valorar de forma cuantitativa la progresión de la enfermedad en el momento del diagnóstico y supervisar la pérdida de función a lo largo del tiempo7
- Cada área funcional se puntúa en una escala del 3 (totalmente normal) al 0 (muy deteriorado)7
- La mayor puntuación posible que se obtiene cuando se evalúan la función motora y el lenguaje es de 68
El desempeño funcional se valora del modo siguiente:7,8,10
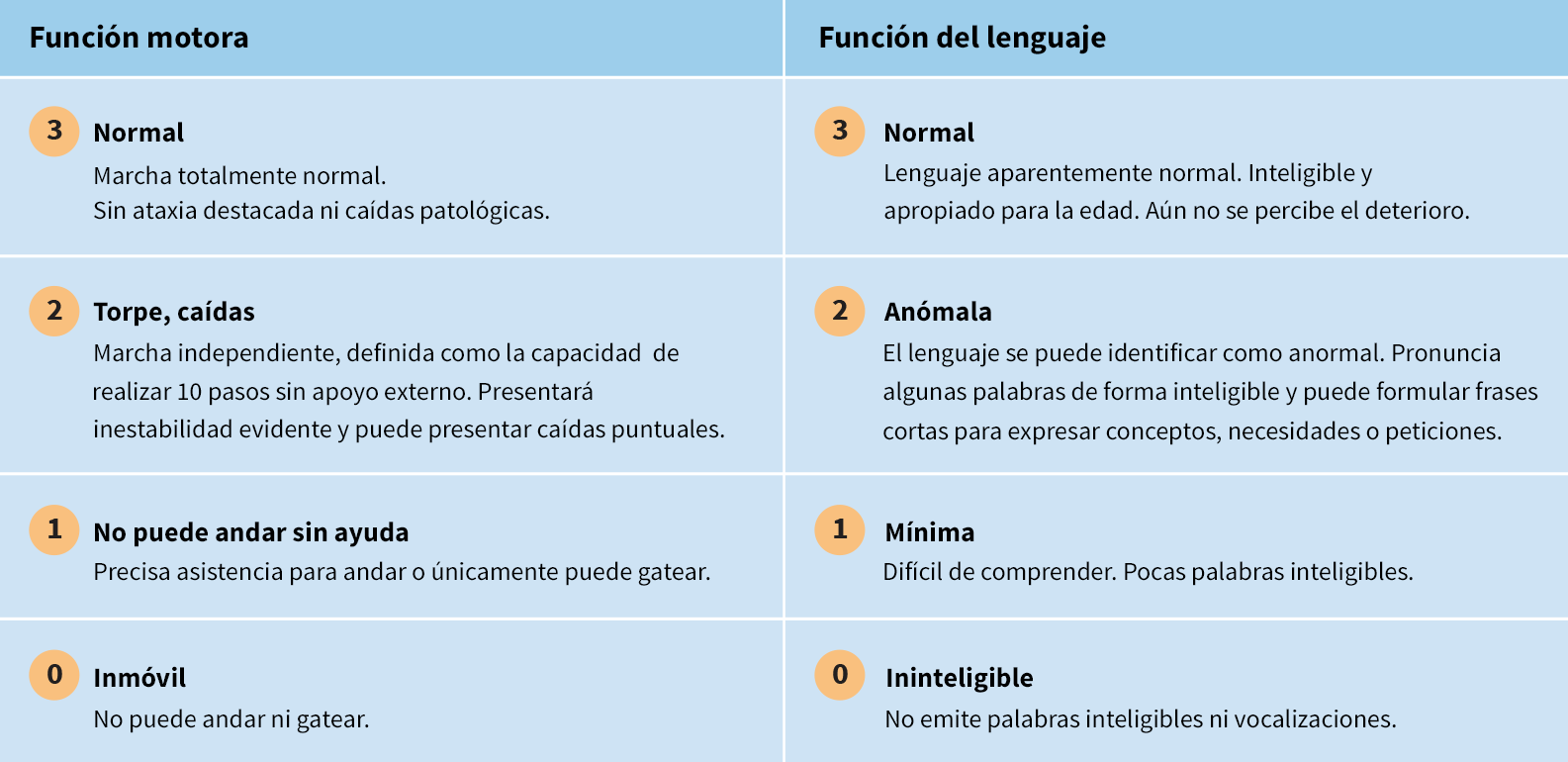
Además de las evaluaciones motoras y del lenguaje, también hay dominios funcionales (cada uno con una escala de 3 puntos) para evaluar la visión y la frecuencia de las convulsiones.7
*Algunos niños puede que nunca presenten actividad motora normal y/o desarrollo del lenguaje. En estos casos, el mejor rendimiento alcanzado por el niño se considera normal. Cuando este nivel de desempeño empeora perceptiblemente, el niño recibe una puntuación de 2 para la función motora y desarrollo del lenguaje ligeramente anormales.
Dr. Alfried Kohlschuetter, MD
Profesor de Pediatría
Departamento de Pediatría
Centro Médico Universitario Hamburg-Eppendorf
Hamburgo, Alemania
Profesor de Pediatría
Departamento de Pediatría
Centro Médico Universitario Hamburg-Eppendorf
Hamburgo, Alemania
Conozca cómo se creó la escala de valoración clínica de la CLN2 para ayudar a los médicos a realizar un seguimiento cuantitativo de la progresión de la enfermedad y conocer más sobre la importancia del papel de los cuidadores en la facilitación de información sobre la progresión de la enfermedad al realizar un seguimiento y una vigilancia sistemáticas de las funciones de los niños con CLN2.
CLN2: una carrera contrarreloj
La mayoría de niños con CLN2 presenta una pérdida consistente de función motora y del lenguaje, tal como se determina en la escala de puntuación clínica de la CLN28

Datos de estudios longitudinales de 58 sujetos con CLN2 del registro DEM-CHILD. IC = intervalo de confianza.
- Edad promedio de aparición de las convulsiones: ~3 años
- Edad promedio del diagnóstico de CLN2: ~5 años
- Pérdida promedio por año: 2 puntos
- Edad promedio de muerte: 10,1 años
Dada la rápida progresión, 2 años hasta el diagnóstico es demasiado tiempo.
Bibliografía: 1. Schulz A, et al. NCL diseases — clinical perspectives. Biochim Biophys Acta. 2013;1832:1801 -1806. 2. Mole SE, Williams RE. Neuronal ceroid-lipofuscinoses. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReview® [internet]. Seattle, WA: University of Washington;1993 -2016. 3. Mole SE et al. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics 2005;6:107 -126. 4. Chang M et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom:Oxford University Press; 2011:80 -109. 5. Pérez-Poyato MS et al. Late Infantile Neuronal Ceroid Lipofuscinosis: Mutations in the CLN2 Gene and Clinical Course in Spanish Patients. J Child Neurol 2013;28:470 -478. 6. Worgall S et al. Treatment of Late Infantile Neuronal Ceroid Lipofuscinosis by CNS Administration of a Serotype 2 Adeno-Associated Virus Expressing CLN2 cDNA. Hum Gene Ther 2008;19:463 -474. 7. Steinfeld R et al. Late Infantile Neuronal Ceroid Lipofuscinosis: Quantitative Description of the Clinical Course in Patients With CLN2 Mutations. Am J Med Genet 2002;112:347 -354. 8. Nickel M et al. Disease characteristics and progression in patients with late-infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease: an observational cohort study. Lancet Child Adolesc Health 2018;2(8):582–590. 9. Fietz M et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): Expert recommendations for early detection and laboratory diagnosis. Mol Genet Metab. 2016;119:160–167. 10. Schulz A et al. Study of Intraventricular Cerliponase Alfa for CLN2 Disease. N Engl J Med. 2018;378:1898–907.
