 Español
Español
 Español
Español UK (English)
UK (English) Deutsch
Deutsch Italiano
Italiano Türkçe
Türkçe Русский
Русский Français
Français
Forget your password?
Necesidad de diagnóstico precoz

Necesidad de diagnóstico precoz
Los signos y síntomas de CLN2 van más allá de la epilepsia
Sin embargo, a menudo se diagnostica epilepsia a los niños sin pruebas adicionales para detectar CLN2
- La edad promedio del diagnóstico son 5 años, habiéndose ya establecido el declive neurodegenerativo de la CLN21
- El diagnóstico de CLN2 puede retrasarse debido a:1
- Síntomas iniciales inespecíficos
- La progresión de la CLN2 puede confundirse con efectos secundarios de los fármacos antiepilépticos
- El diagnóstico precoz facilita el acceso a cuidados específicos de la CLN2 y a apoyo social, que pueden tener una repercusión positiva sobre la calidad de vida del niño y la familia1,2
- Obtener un diagnóstico precoz de la CLN2 es fundamental para la sensibilización de los padres como portadores genéticos y refuerza la importancia de la planificación familiar3
- Los retrasos en el diagnóstico puede ir acompañados de evaluaciones exhaustivas y diagnósticos erróneos antes de confirmar definitivamente el diagnóstico2,4
El diagnóstico de CLN2 se retrasa un promedio de 2 años desde la primera convulsión detectada a la edad de 3 años.
A menudo, la CLN2 se diagnostica incorrectamente al principio, lo que retrasa la obtención de un diagnóstico preciso
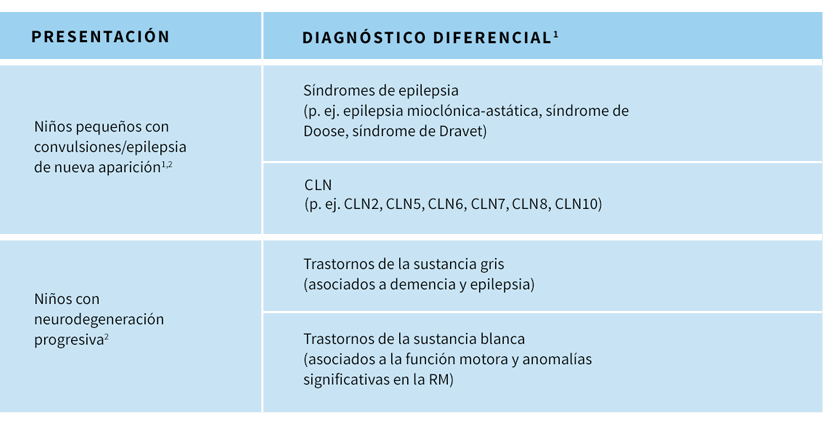
El diagnóstico diferencial de la CLN2 varía en función del estadio de progresión de la enfermedad.En periodos tempranos de progresión de la CLN23,4
- Debido a que las convulsiones y la mioclonía son los síntomas más destacados, a menudo se sospecha de síndromes dónde la mioclonía es frecuente, p. ej. epilepsia focal, síndromes epilépticos mioclónicos/epilepsias astáticas mioclónicas, como el síndrome de Doose, síndrome de Dravet, síndrome de Lennox-Gastaut u otros síndromes epilépticos
En periodos tardíos de progresión de la CLN2:3
- A medida que la enfermedad progresa y la regresión psicomotora y la pérdida funcional se hacen más evidentes, puede sospecharse de otros trastornos neurológicos pediátricos progresivos, como inflamación/infecciones, tumores, trastornos mitocondriales, otros trastornos de depósito lisosómico, incluyendo otros tipos de CLN
References: 1. Fietz M et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): Expert recommendations for early detection and laboratory diagnosis. Mol Genet Metab. 2016; 119:160–167 2. Williams RE et al. Management Strategies for CLN2 Disease. Pediatr Neurol 2017;69:102–112. 3. Chang M et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 4. Pérez-Poyato MS et al. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. J Child Neurol. 2013;28:470-478. 5. Mole SE, Williams RE, and Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126.
