 Español
Español
 Español
Español UK (English)
UK (English) Deutsch
Deutsch Italiano
Italiano Türkçe
Türkçe Русский
Русский Français
Français
Forget your password?
Fisiopatología

Fisiopatología
La deficiencia de la enzima TPP1 provoca neurodegeneración en niños con CLN2.1
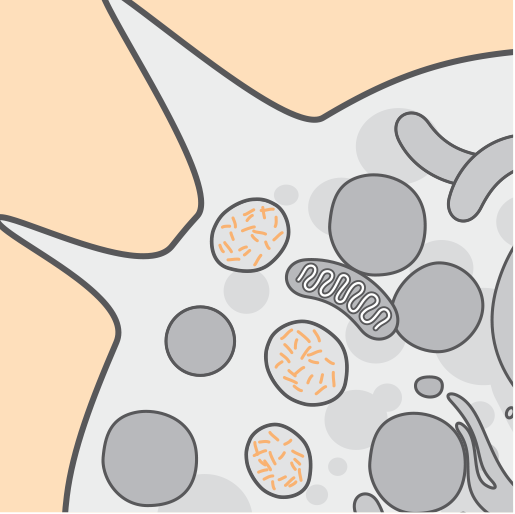
Las mutaciones en el gen CLN2 / TPP1 dan como resultado una actividad deficiente de la enzima tripeptidil-peptidasa 1 (TPP1). TPP1 es una proteasa que escinde los tripéptidos N-terminales de los sustratos en los lisosomas.3-5
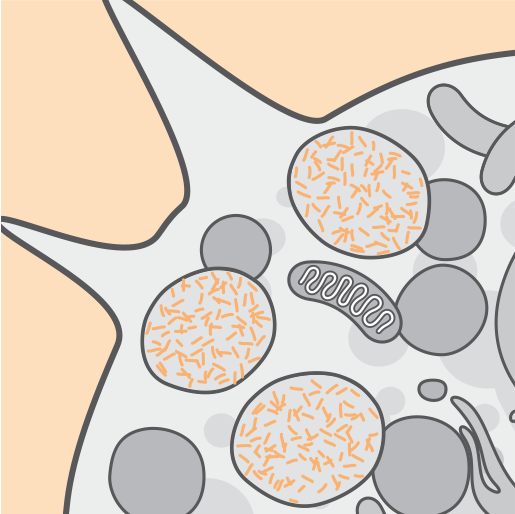
La ausencia o actividad reducida de la enzima TPP1 está asociada. con una acumulación de material lipopigmento autofluorescente lisosómico2,4.
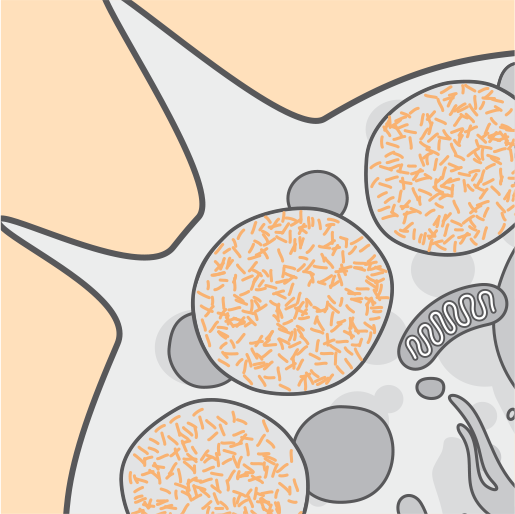
Con el tiempo, disfunción celular, celular.Ocurren muerte y atrofia.3,6
La acumulación continua en los lisosomas y la progresión de los síntomas devastadores se observan a lo largo del curso de la enfermedad CLN2.
Bibliografía: 1. Schulz A et al. NCL diseases — clinical perspectives. Biochim Biophys Acta 2013;1832:1801–1806. 2. Chang M et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom:Oxford University Press; 2011:80–109. 3. Mole SE, Williams RE. Neuronal ceroid-lipofuscinoses. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReview® [internet]. Seattle, WA: University of Washington;1993–2016. 4. Mole SE et al. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics 2005;6:107–126. 5. Fietz M et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): Expert recommendations for early detection and laboratory diagnosis. Mol Genet Metab. 2016;119:160–167. 6. Haltia M. The neuronal ceroid-lipofuscinoses: From past to present. Biochim Biophys Acta 2006;1762:850–856.
