Ce site s'adresse aux professionnels de la santé uniquement. Veuillez cliquer ici si vous n'êtes pas un professionnel de la santé.
 Français
Français
 UK (English)
UK (English) Español
Español Italiano
Italiano Deutsch
Deutsch Türkçe
Türkçe Русский
Русский
Forget your password?
Histoire naturelle

Histoire naturelle
L'évolution de la CLN2 est rapide et dévastatrice — les symptômes et les pertes fonctionnelles s'aggravent avec l'âge
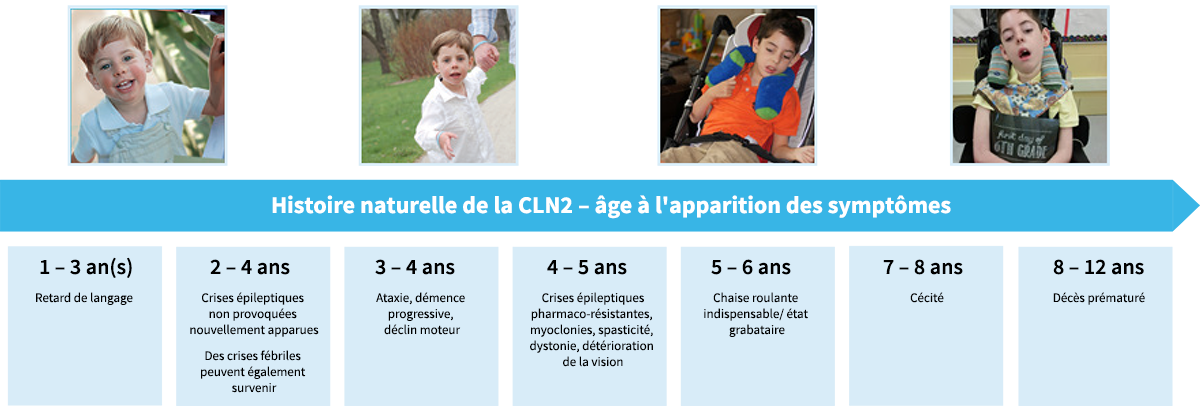
Chronologie de l'apparition des symptômes et des pertes fonctionnelles dans la forme classique de la CLN21-6

Les tranches d'âge indiquées sont des moyennes pour le phénotype infantile tardif classique. Pour les phénotypes atypiques de la CLN2, l'âge d'apparition, le rythme de progression et les manifestations de la maladie peuvent être variables.
Âge de 0 à 2 ans
- L'accumulation des lipopigments dans les lysosomes débute vraisemblablement au stade prénatal ; mais les enfants semblent se développer normalement jusqu'à l'apparition des premiers symptômes2
- Des observations cliniques récentes suggèrent que le premier symptôme est, dans la majorité des cas, un retard de langage précoce7
- Dans une minorité de cas, d'autres retards de développement, des difficultés motrices ou un ataxie peuvent constituer le premier signe4,5
- Ces signes surviennent souvent conjointement avec un retard de langage
Âge de 2 à 4 ans
- Bien que le retard de langage puisse apparaître plus tôt, ce sont les crises d'épilepsie (survenant vers l'âge d'environ 3 ans) qui incitent le plus souvent les parents à consulter pour la CLN22,8
- Les crises peuvent être de divers types, par exemple des crises généralisées tonico-cloniques, des crises d'absence, ou des crises myocloniques, atoniques, cloniques et toniques2,4
- Bien que les crises soient le plus souvent non provoquées, des crises fébriles peuvent également se produire4
- L'ataxie se manifeste souvent à ce stade de la maladie5
Âge de 4 à 7 ans
- Les symptômes s'aggravent avec l'âge et s'accompagnent d'une perte considérable des aptitudes acquises antérieurement. À cet âge, les enfants atteints de CLN2 présentent généralement :1,2,8,9
- une régression du langage
- des crises myocloniques, épileptiques et non épileptiques
- une altération sévère des fonctions motrices avec, à terme, une perte des mouvements volontaires
- des mouvements anormaux de type myoclonie, spasticité, dystonie et chorée
- une détérioration de la vision, conduisant à une cécité vers l'âge de 6 ou 7 ans
- Finalement, une perte complète du langage et de l'ambulation est observée, contraignant les enfants à la chaise roulante ou à rester alités et totalement dépendants de leurs aidants2
Âge de 8 à 12 ans
- La CLN2 est caractérisée par une mortalité prématurée et les enfants survivent rarement au-delà de la prime adolescence1
La cécité est un symptôme précoce pour les autres CLN, mais elle survient plus tardivement dans la CLN2.6,9
Dr Paul Gissen, MBChB, PhD, FRCPCH
Collège universitaire de Londres
Hôpital des enfants de Great Ormond Street
NHS Foundation Trust
Collège universitaire de Londres
Hôpital des enfants de Great Ormond Street
NHS Foundation Trust
Présentation de la nature prévisible de la CLN2 et moment d'apparition des différents symptômes au cours de la vie des enfants affectés.
L'échelle d'évaluation clinique de la CLN2 est un outil efficace pour évaluer la progression de la maladie dans 2 domaines fonctionnels majeurs : les aptitudes motrices et les aptitudes au langage
Ce système de notation standardisé peut être utilisé pour évaluer quantitativement la progression de la maladie lors du diagnostic et suivre l'évolution des pertes fonctionnelles au cours du temps6
- Chaque domaine fonctionnel est évalué sur une échelle allant de 3 (globalement normal) à 0 (altération profonde)6
- Le score le plus élevé possible lors de l'évaluation des fonctions motrices et du langage est de 67
Les scores d'évaluation des aptitudes fonctionnelles sont définis comme suit :6
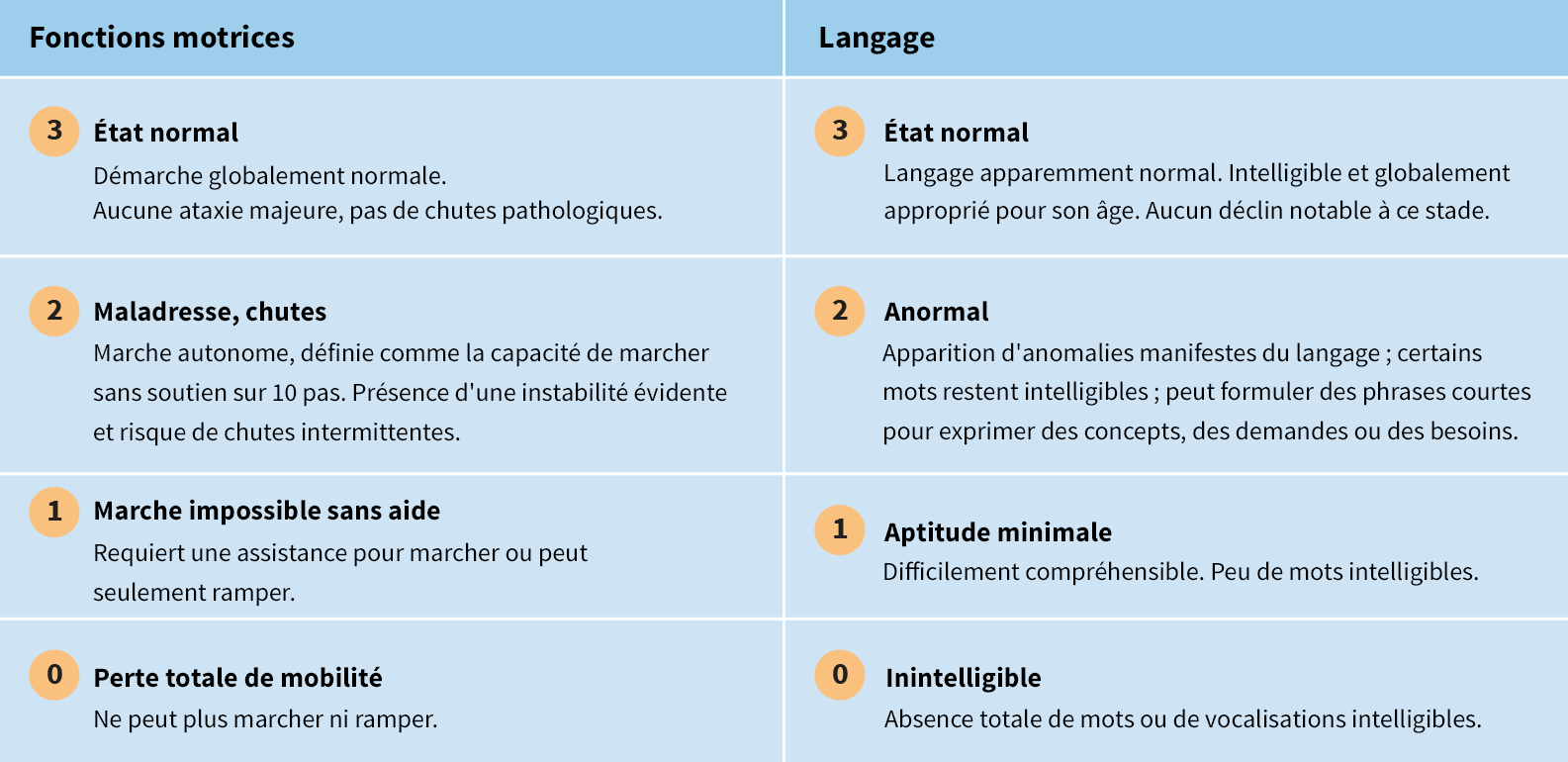
Outre les évaluations des fonctions motrices et du langage, d'autres domaines fonctionnels (mesurés chacun sur une échelle à 3 points) permettent également d'évaluer la vision et la fréquence des crises.6
* Chez certains enfants, il n'y a jamais eu de fonctions motrices normales et/ou d'acquisition normale du langage. Dans ce cas, le niveau de performance maximal atteint par l'enfant est considéré comme normal. Dès lors qu'une détérioration visible des performances est constatée, un score de 2 est attribué à l'enfant pour signifier une légère anomalie des fonctions motrices et/ou de l'acquisition du langage.
Dr Alfried Kohlschuetter, MD
Professeur en pédiatrie
Département de pédiatrie
Centre médical universitaire Hamburg-Eppendorf
Hambourg, Allemagne
Professeur en pédiatrie
Département de pédiatrie
Centre médical universitaire Hamburg-Eppendorf
Hambourg, Allemagne
Cette vidéo explique comment l'échelle d'évaluation clinique de la CLN2 a été créée pour aider les médecins à assurer un suivi quantitatif de la progression de la maladie, et décrit le rôle important joué par les aidants pour fournir des indications sur la progression de la maladie grâce à l'attention prêtée quotidiennement à l'état fonctionnel de l'enfant atteint de CLN2.
La CLN2 est une course contre le temps
La majorité des enfants atteints de CLN2 connaissent une régression constante des fonctions motrices et du langage, comme mesurée via l'échelle d'évaluation clinique de la CLN27

Données longitudinales portant sur 58 patients atteints de CLN2 du registre DEM-CHILD. IC : intervalle de confiance.
- Âge moyen lors de la première crise : ~3 ans
- Âge moyen lors du diagnostic de la CLN2 : ~5 ans
- Perte moyenne par année : 2 points
- Âge moyen lors du décès : 10,1 ans
Étant donné la progression rapide de la maladie, un délai de 2 ans avant le diagnostic est trop long.
Références : 1. Mole SE, Williams RE, and Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126. 2. Chang M, Cooper JD, Davidson BL, et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 3. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases – clinical perspectives. Biochimica et Biophysica Acta. 2013;1832:1801–1806. 4. Pérez-Poyato MS, Marfa MP, Abizanda IF, et al. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. J Child Neurol. 2013;28:470-478. 5. Worgall S, Sondhi D, Hackett NR, et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum Gene Ther. 2008;19:463-474. 6. Steinfeld R, Heim P, von Gregory H, et al. Late infantile neuronal ceroid lipofuscinosis: quantitative description of the clinical course in patients with CLN2 mutations. Am J Med Genet. 2002;112:347-354. 7. Nickel M, Jacoby D, Lezius S, et al. Natural history of CLN2 disease: quantitative assessment of disease characteristics and rate of progression. Poster session presented at: The 12th Annual WORLD Symposium; February – March 2016; San Diego, CA. 8. Schulz A, Cohen-Pfeffer JL, Crystal R, et al. Neuronal ceroid lipofuscinosis-2 (CLN2) disorder, a type of Batten disease caused by TPP1 enzyme deficiency: current knowledge of the natural history from international experts. Poster session presented at: The Society for the Study of Inborn Errors of Metabolism (SSIEM) Annual Symposium; September 2015; Lyon, France. 9. Mole SE, Williams RE. Neuronal ceroid-lipofuscinoses. 2001 Oct 10 [Updated 2013 Aug 1]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [internet]. Seattle, WA: University of Washington; 1993-2016.
