Ce site s'adresse aux professionnels de la santé uniquement. Veuillez cliquer ici si vous n'êtes pas un professionnel de la santé.
 Français
Français
 UK (English)
UK (English) Español
Español Italiano
Italiano Deutsch
Deutsch Türkçe
Türkçe Русский
Русский
Forget your password?
Physiopathologie

Physiopathologie
Le déficit en enzyme TPP1 entraîne une neurodégénérescence chez les enfants atteints de CLN21
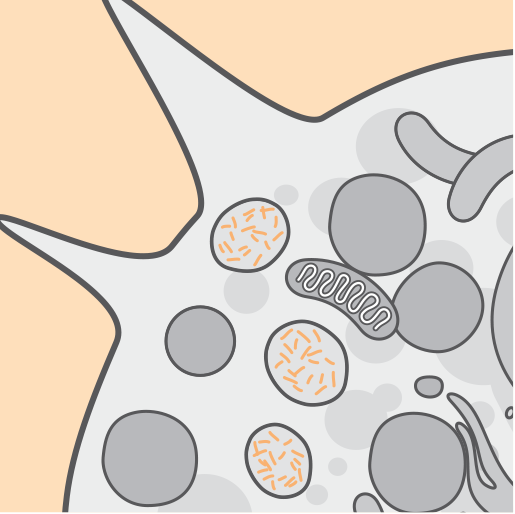
Les mutations du gène CLN2/TPP1 engendrent un déficit de l'activité de l'enzyme tripeptidyl-peptidase 1 (TPP1). La TPP1 est une protéase qui clive les tripeptides de l'extrémité N-terminale de substrats dans les lysosomes.1,2
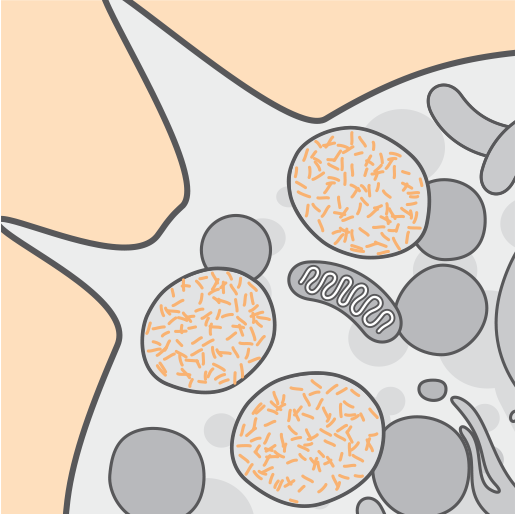
L'absence ou l'activité réduite de l'enzyme TPP1 conduit à l'accumulation d'un lipopigment autofluorescent dans les lysosomes.2,3
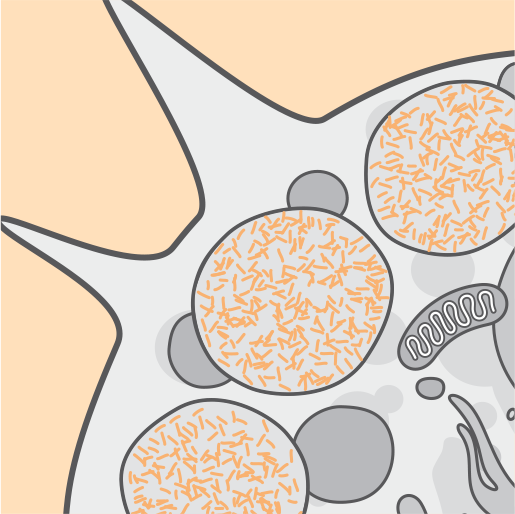
Cela provoque, à terme, un dysfonctionnement cellulaire, la mort cellulaire et une atrophie.3,4
L'accumulation continue dans les lysosomes détériore les cellules et les symptômes dévastateurs de la CLN2 deviennent perceptibles.
Références : 1. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases – clinical perspectives. Biochimica et Biophysica Acta. 2013;1832:1801-1806. 2. Mole SE, Williams RE, and Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126. 3. Chang M, Cooper JD, Davidson BL, et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 4. Haltia M. The neuronal ceroid-lipofuscinoses: from past to present. Biochimica et Biophysica Acta. 2006;1762:850-856.
