Ce site s'adresse aux professionnels de la santé uniquement. Veuillez cliquer ici si vous n'êtes pas un professionnel de la santé.
 Français
Français
 UK (English)
UK (English) Español
Español Italiano
Italiano Deutsch
Deutsch Türkçe
Türkçe Русский
Русский
Forget your password?
Nécessité d’un diagnostic précoce

Nécessité d'un diagnostic précoce
Les signes et symptômes de la CLN2 s'étendent au-delà de l'épilepsie
Pourtant, un simple diagnostic d'épilepsie est souvent établi pour ces enfants, sans autres examens recherchant une CLN2
- L'âge moyen au moment du diagnostic est de 5 ans, c'est-à-dire une fois le déclin neurodégénératif associé à la CLN2 bien avancé1
- Le diagnostic de la CLN2 peut être retardé en raison :1
- du caractère non spécifique des symptômes initiaux
- d'une confusion entre la progression de la CLN2 et les effets indésirables des anti-épileptiques
- Un diagnostic précoce permet l'accès à des soins cliniques spécifiques à la CLN2 et à un soutien social, qui peuvent avoir un impact positif sur l'évolution de la maladie et la qualité de vie de l'enfant et de sa famille1,2
- Un diagnostic plus précoce de la CLN2 est essentiel pour sensibiliser les parents au risque d'être des porteurs génétiques de la maladie et pour soutenir l'importance de la planification familiale3
- Des retards de diagnostic peuvent être associés à de nombreux examens épuisants et à l'établissement de diagnostics erronés, avant la pose du diagnostic définitif2,4
Le diagnostic de la CLN2 est établi avec un retard moyen de 2 ans après la première crise observée à l'âge de 3 ans.
La CLN2 est souvent mal diagnostiquée au stade précoce, retardant l'établissement du diagnostic approprié
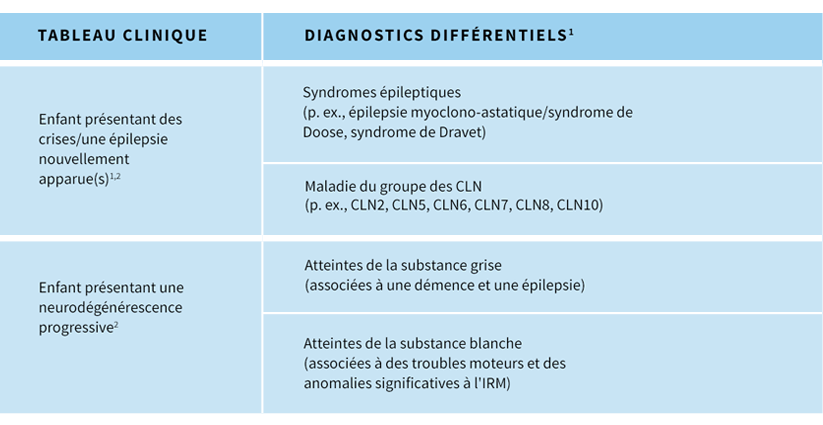
Le diagnostic différentiel de la CLN2 varie selon le stade de progression de la maladie.Stade précoce de la progression de la CLN2 :3,5
- Les crises d'épilepsie et les myoclonies constituant des symptômes prédominants, il est fréquent de suspecter des syndromes qui sont généralement associés à des myoclonies (p. ex., épilepsie focale ; syndromes d'épilepsie myoclonique/épilepsies myoclono-astatiques du type syndrome de Doose, syndrome de Dravet, syndrome de Lennox-Gastaut ; ou autres syndromes épileptiques)
Stade plus avancé de progression de la CLN2 :3
- À mesure que la maladie progresse et que de la régression psychomotrice et la perte fonctionnelle s'amplifient, d'autres maladies cérébrales pédiatriques progressives peuvent être suspectées (p. ex., inflammation/infections, tumeurs, maladies mitochondriales et autres maladies de surcharge lysosomale, y compris les autres types de CLN)
Références : 1. Fietz M, Giugliani R, AlSayed M, et al. Expert recommendations for the laboratory diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): diagnostic algorithm and best practice guidelines for a timely diagnosis. Poster session presented at: The 12th Annual WORLD Symposium; February – March 2016; San Diego, CA. 2. Williams RE, Adams HR, Blohm M, et al. Expert opinion on the management of CLN2 disease. Poster session presented at: The 12th Annual WORLD Symposium; February – March 2016; San Diego, CA. 3. Chang M, Cooper JD, Davidson BL, et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 4. Pérez-Poyato MS, Marfa MP, Abizanda IF, et al. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. J Child Neurol. 2013;28:470-478. 5. Mole SE, Williams RE, and Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126.
