 Español
Español UK (English)
UK (English) Deutsch
Deutsch Italiano
Italiano Русский
Русский Français
Français
Forget your password?
História natural

História natural
Na CLN2, as crianças apresentam perda importante de funções à medida que os sintomas aumentam com a idade
Linha do tempo da manifestação de sintomas da doença CLN2 clássica e perda da função1-6

A demonstração da faixa etária são médias para o fenótipo clássico infantil tardio. Os fenótipos atípicos da doença CLN2 podem variar em idade de início, taxa de progressão e manifestações da doença.
0 a 2 anos
- O acúmulo lisossômico de lipopigmentos começa, provavelmente, no período pré-natal, ainda que as crianças pareçam se desenvolver normalmente até o início da apresentação dos sintomas1
- O primeiro sintoma na maioria dos casos é o atraso precoce de linguagem, conforme mostrado nos últimos achados clínicos7
- Numa minoria de casos, outros atrasos no desenvolvimento, dificuldades motoras ou ataxia podem ser o primeiro sinal4,5
- Muitas vezes eles podem ocorrer em conjunto com o atraso da linguagem
2 a 4 anos
- Apesar da possibilidade do atraso da linguagem aparecer mais cedo, as convulsões (que ocorrem em torno dos 3 anos de idade) representam o sintoma que mais leva os pais a procurarem atendimento médico para a doença CLN22,8
- Pode-se observar vários tipos de convulsões, incluindo as tônico-clônicas generalizadas, ausência, mioclônicas, atônicas, clônicas e tônicas2,4
- Embora as crises sejam, na maioria das vezes, não provocadas, podem ocorrer convulsões febris4
- O aparecimento da ataxia muitas vezes ocorre durante este estágio da doença5
4 a 7 anos
- Os sintomas se acumulam com a idade e há uma perda significativa de habilidades adquiridas. Durante esta faixa etária, as crianças com a doença CLN2 normalmente apresentam1,2,8,9:
- Regressão da linguagem
- Crises mioclônicas, tanto epiléticas quanto não epilépticas
- Grave comprometimento da função motora, com eventual perda de movimentos voluntários
- Transtornos do movimento, tais como mioclonia, espasticidade, distonia e coreia
- Deterioração visual que leva à cegueira aos 6 ou 7 anos
- Eventualmente há uma perda completa da linguagem e locomoção, e as crianças tornam-se dependentes de cadeira de rodas, acamadas e completamente dependentes de cuidadores2
8 a 12 anos
- A doença CLN2 é caracterizada pela morte prematura e a maioria das crianças raramente sobrevive após o início da adolescência1
A cegueira é um sintoma precoce em outras LCNs, mas ocorre na fase posterior da doença CLN26,9

Dr. Paul Gissen, MBChB, PhD, FRCPCH
University College London
Hospital Infantil Great Ormond Street
NHS Foundation Trust
University College London
Hospital Infantil Great Ormond Street
NHS Foundation Trust
Uma visão geral da natureza previsível da doença CLN2 e quando os sintomas se desenvolvem ao longo da vida das crianças afetadas.
A Escala de Avaliação Clínica da Doença CLN2 é uma maneira eficiente de avaliar a progressão da doença em duas grandes áreas funcionais: habilidade motora e de linguagem
O sistema de pontuação padronizado pode ser utilizado para avaliar quantitativamente a progressão da doença e controlar a perda de função ao longo do tempo6
- Cada área funcional é pontuada em uma escala de 3 (normal) a 0 (prejudicada profundamente)10
- A maior pontuação possível ao avaliar a função motora e de linguagem é 67
A capacidade de desempenho funcional é classificada da seguinte forma6:
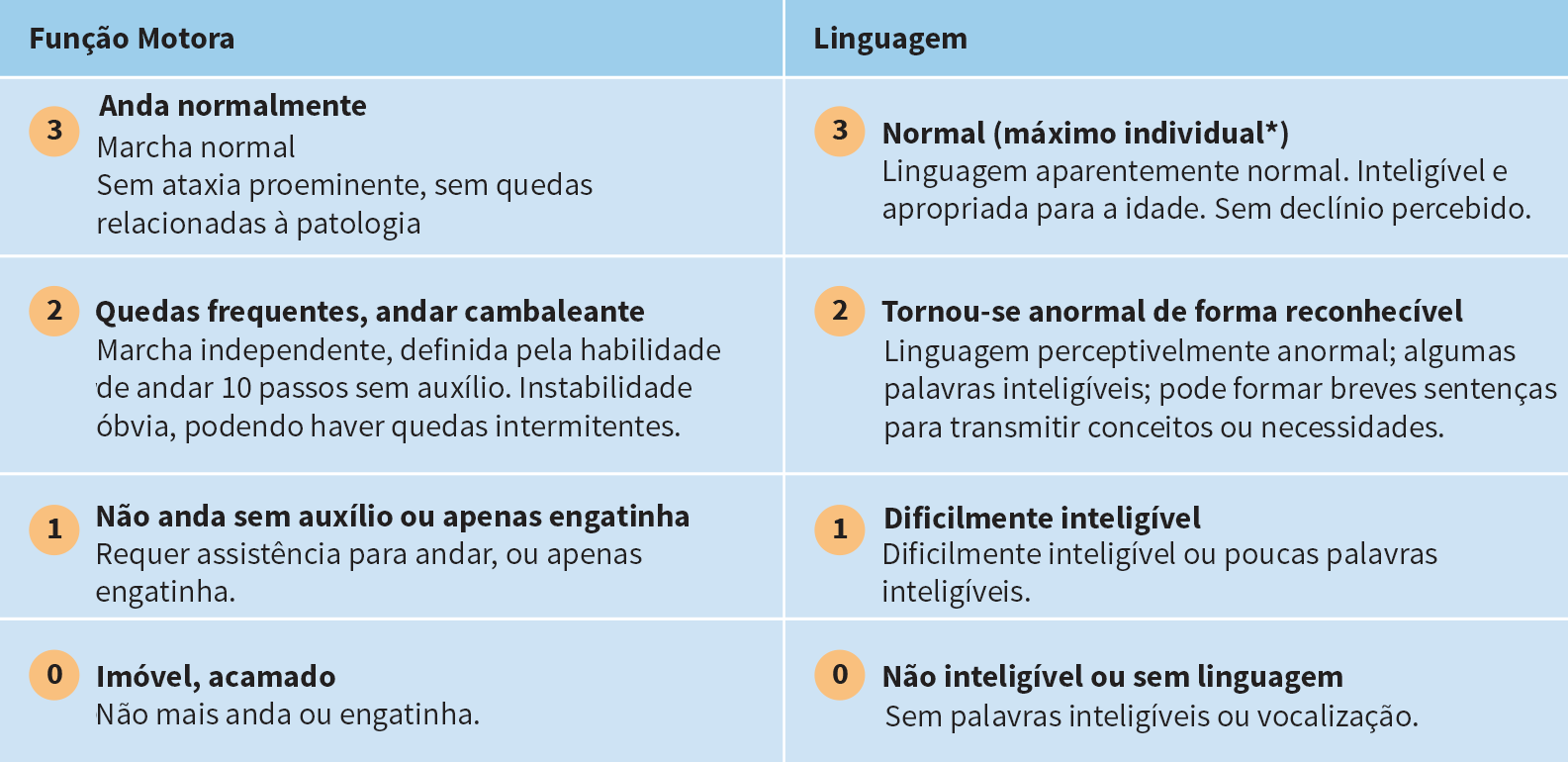
Além das avaliações da função motora e de linguagem, há também domínios funcionais (cada um com uma escala de 3 pontos) para avaliar a visão e a frequência das convulsões.6
*Em algumas crianças, o desenvolvimento motor normal e/ou da linguagem nunca esteve presente. Em tais casos, o melhor desempenho obtido pela criança foi considerado normal. Quando esse nível de desempenho tornou-se reconhecidamente pior, a criança foi avaliada como 2 para a função motora e/ou desenvolvimento da linguagem ligeiramente anormal.
Dr Alfried Kohlschuetter, Médico
Professor de Pediatria
Departamento de Pediatria
University Medical Center Hamburg-Eppendorf
Hamburgo, Alemanha
Professor de Pediatria
Departamento de Pediatria
University Medical Center Hamburg-Eppendorf
Hamburgo, Alemanha
Ouça como a Escala de Avaliação Clínica da Doença CLN2 foi criada para ajudar os médicos a controlar quantitativamente a progressão da doença e saiba mais sobre o importante papel que os cuidadores desempenham na prestação de informações sobre a progressão da doença, conforme acompanham e monitoram as funções de rotina nos seus filhos com a doença CLN2.
A doença CLN2 é uma corrida contra o tempo
A maioria das crianças com a doença CLN2 apresentam uma perda consistente da função motora e da linguagem, tal como medido pela Escala de Avaliação Clínica da Doença CLN27

Dados longitudinais de 58 indivíduos com doença CLN2 no registro DEM-CHILD. IC intervalo de confiança
- Idade média da primeira convulsão: ~3 anos
- Idade média do diagnóstico da doença CLN2: ~5 anos
- Perda média por ano: 2 pontos
- Idade média da morte: 10,1 anos
Devido a uma rápida taxa de progressão, o prazo de dois anos para o diagnóstico é muito longo.
Referencias: 1. Mole SE, Williams RE, and Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6:107-126. 2. Chang M, Cooper JD, Davidson BL, et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109. 3. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases – clinical perspectives. Biochimica et Biophysica Acta. 2013;1832:1801–1806. 4. Pérez-Poyato MS, Marfa MP, Abizanda IF, et al. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. J Child Neurol. 2013;28:470-478. 5. Worgall S, Sondhi D, Hackett NR, et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum Gene Ther. 2008;19:463-474. 6. Steinfeld R, Heim P, von Gregory H, et al. Late infantile neuronal ceroid lipofuscinosis: quantitative description of the clinical course in patients with CLN2 mutations. Am J Med Genet. 2002;112:347-354. 7. Nickel M, Jacoby D, Lezius S, et al. Natural history of CLN2 disease: quantitative assessment of disease characteristics and rate of progression. Poster session presented at: The 12th Annual WORLD Symposium; February – March 2016; San Diego, CA. 8. Schulz A, Cohen-Pfeffer JL, Crystal R, et al. Neuronal ceroid lipofuscinosis-2 (CLN2) disorder, a type of Batten disease caused by TPP1 enzyme deficiency: current knowledge of the natural history from international experts. Poster session presented at: The Society for the Study of Inborn Errors of Metabolism (SSIEM) Annual Symposium; September 2015; Lyon, France. 9. Mole SE, Williams RE. Neuronal ceroid-lipofuscinoses. 2001 Oct 10 [Updated 2013 Aug 1]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [internet]. Seattle, WA: University of Washington; 1993-2016.
